Prucalopride
57. Asparlas(Calaspargase Pegol )
12月20日,FDA批准了施维雅的Asparlas,作为多药化疗方案的一个成分,用于1个月至21岁的急性淋巴细胞白血病(ALL)患者治疗。Asparlas含有L-天冬酰胺酶(L-天冬酰胺酰胺水解酶),而L-天冬酰胺酶可催化L-天冬酰胺转化为天冬氨酸和氨。ASPARLAS的药理作用被认为是通过降低血浆中天冬酰胺的含量,而间接地杀灭高度依赖外源性天冬酰胺的白血病细胞[96]。因为本品是在培门冬酶(Pegaspargase)的基础上发展起来的,而培门冬酶用于ALL,已经是临床证据非常充分的一线疗法[97]。因此本品的疗效仅需证明每三周一次静脉注射本品2500U/m2,可实现并维持天冬酰胺酶的活性(NSAA)高于0.1U/mL。试验共有124例B细胞系急性淋巴细胞白血病(ALL)患者入组,通过药代动力学检测,123名患者在第6周,第12周,第18周,第24周和第30周的NSAA均维持在0.1U/mL以上[96]。尽管也本品是门冬酰胺酶类药物,但给药时间间隔更长,具有明显的临床优势。
58. Ultomiris (Ravulizumab)
12月21日,FDA批准了亚力兄弟公司的Ravulizumab,用于阵发性睡眠性血红蛋白尿症(PNH)治疗。Ravulizumab是一种人源化,以补体5(C5)为靶点的单抗,为依库珠单抗的下一代[98]。FDA批准本品主要是基于一项针对PNH的Ⅲ期临床试验(NCT02946463)的结果,在该试验中,患者基于体重在第一天给予一个负荷剂量,第15天给予维持剂量( 40至60kg体重:负荷剂量2400mg, 维持剂量每8周3000mg;60至100kg体重: 负荷剂量2700mg, 维持剂量每8周3300mg;体重大于100kg的患者,负荷剂量3000mg, 维持剂量每8周3600mg),而依库珠单抗则根据说明给药。经过26周的治疗,本品治疗组患者避免输血率为73.6%,乳酸脱氢酶(LDH)正常化率为53.6%,而依库珠单抗分别只有66.1%和49.4%,达到预设的临床终点,两组患者间疲劳状况并无显著性差异[99]。依库珠单抗是世界上最天价的药物之一,年销售额高达30亿美元,是亚力兄弟公司的摇钱树。随着依库珠单抗的日益走俏,布局C5领域的公司开始增多,Ravulizumab的获批有望让该公司继续保持领先的优势,科睿唯安预测本品在2024年的销售额可达18.98亿美元。
59. Elzonris (Tagraxofusp-erzs)
12月21日,FDA还批准了Stemline Therapeutics公司的Elzonris,用于母细胞性浆细胞样树突状细胞肿瘤(BPDCN)治疗。BPDCN是一种罕见的侵袭性骨髓和血液疾病,可以影响多个器官,包括淋巴结和皮肤,它通常表现为白血病或进展为急性白血病。FDA批准本品是基于一项多中心的单臂临床试验(NCT 02113982)的数据,受试者被分成了两个亚组,亚组一的13名患者均为初治型BPDCN,经过本品的治疗,7名患者(54%)达到完全缓解(CR)或皮肤异常并未表明活动性疾病的完全缓解(CRc);亚组二的15名患者则均为复发或难治性BPDCN,经过本品的治疗,一名患者达到CR,一名患者达到CRc[100]。在本品之前,FDA并批准专门针对BPDCN的药物,因此本品获得了突破性疗法、优先审评和孤儿药三项认定[101]。虽然BPDCN是一种非常罕见的疾病,但本品有多个适应症正在开展临床试验,随着适应症的不断拓宽,本品的市场潜力将不断地展现出来,科睿唯安预测本品在2024年的销售额可达3.29亿美元。
小结与展望
2018年是令人非常兴奋的一年,在这一年里,FDA批准了59个新药。对于患者而言,他们可能盼来了救命的神药,对创新药研发人而言,或许看到了更美好的未来。虽然“59”开创了新纪录,但不是永远的纪录!因为在过去的大半个世纪里,创新药的研发投入一直在增加,创新药研发资源一直在扩大,FDA批准新分子实体数量也在曲线上升,随着新靶点的不断发现和新技术的不断运用,这个纪录必将很快被打破。
虽然中国的制药产业还是以仿制药为主,但在“限制辅助性用药”和“仿制药带量采购”的大背景下,仿制药将不再是我国医药市场的主要增长点。在过去的一年里,NMPA批准了近50个新分子实体,其中还包括5个本土创新药,经过“腾笼换鸟”,很多具有巨大临床需求的新分子实体将有望纳入医保,中国的药品市场正在从“以市场需求为导向”到“以临床需求为导向”的模式转变,而医疗保障也正在“从无到有”到“从有到优”转变。在国家一系列政策的推动下,我国的很多仿制药企将“破釜沉舟”做创新药和创新制剂,相信在不远的将来,FDA批准的新药中,将出现来自中国的产品。
创新药占据了全球70%以上的药品市场,是制药行业利润的主要来源,我国制药企业要做大、做强,必须要从仿制到创新转型。虽然我国的创新药事业起步较晚、研发投入较低,但FDA批准的新药为我们提供了很多低技术的廉价创新案例,虽然PhRMA报告指出,创新药的平均研发成本(算失败项目)高达26亿美元,但并非每一个都那么天价,Tpoxx和Asparlas就是最典型的代表,Tpoxx从研发到上市,没有在人上做过有效性临床试验,而Asparlas仅仅只是基于药动学的数据就获批了……总而言之,他山之石可以攻玉,希望本文能够为广大同仁带来丝毫的帮助!
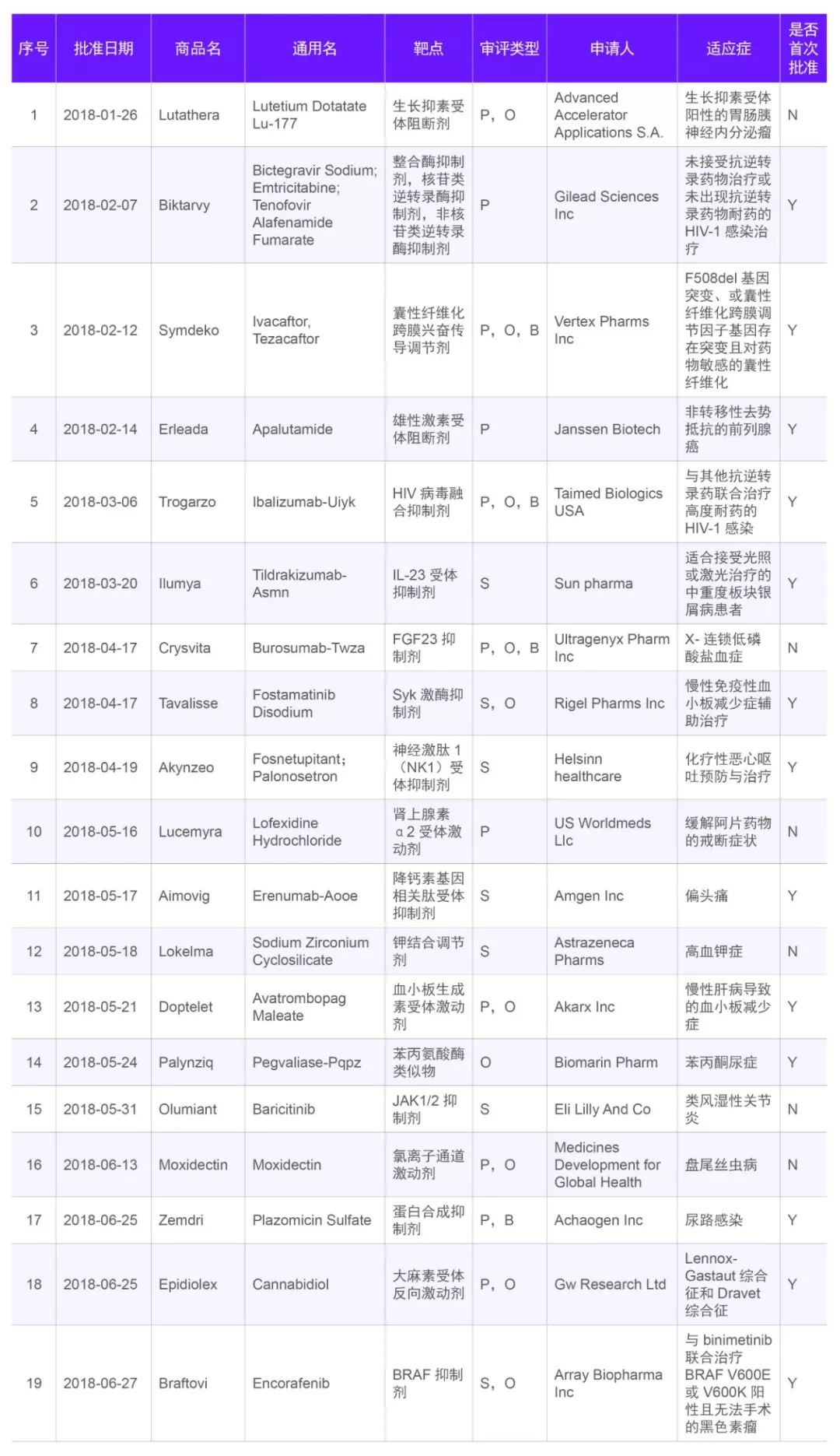
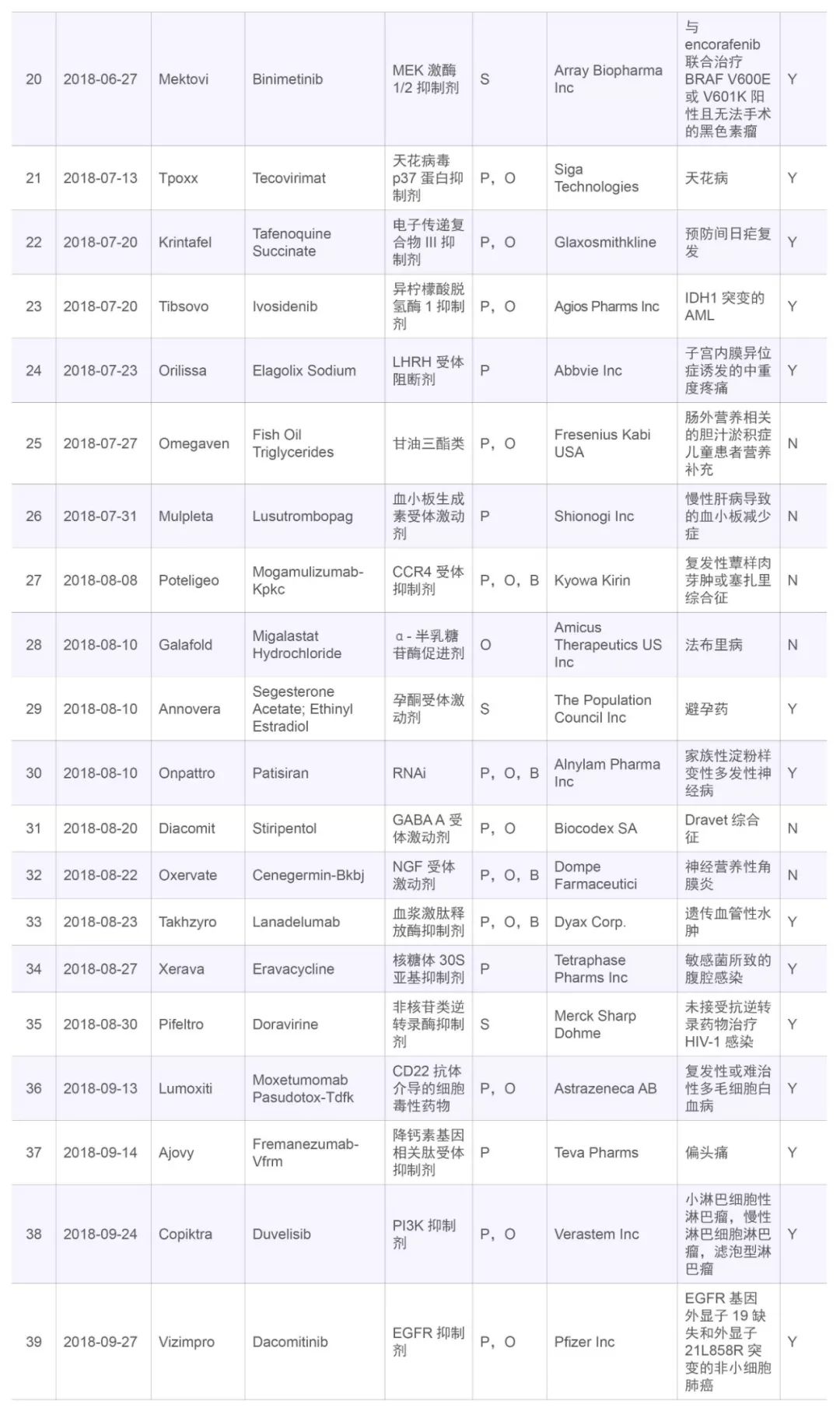
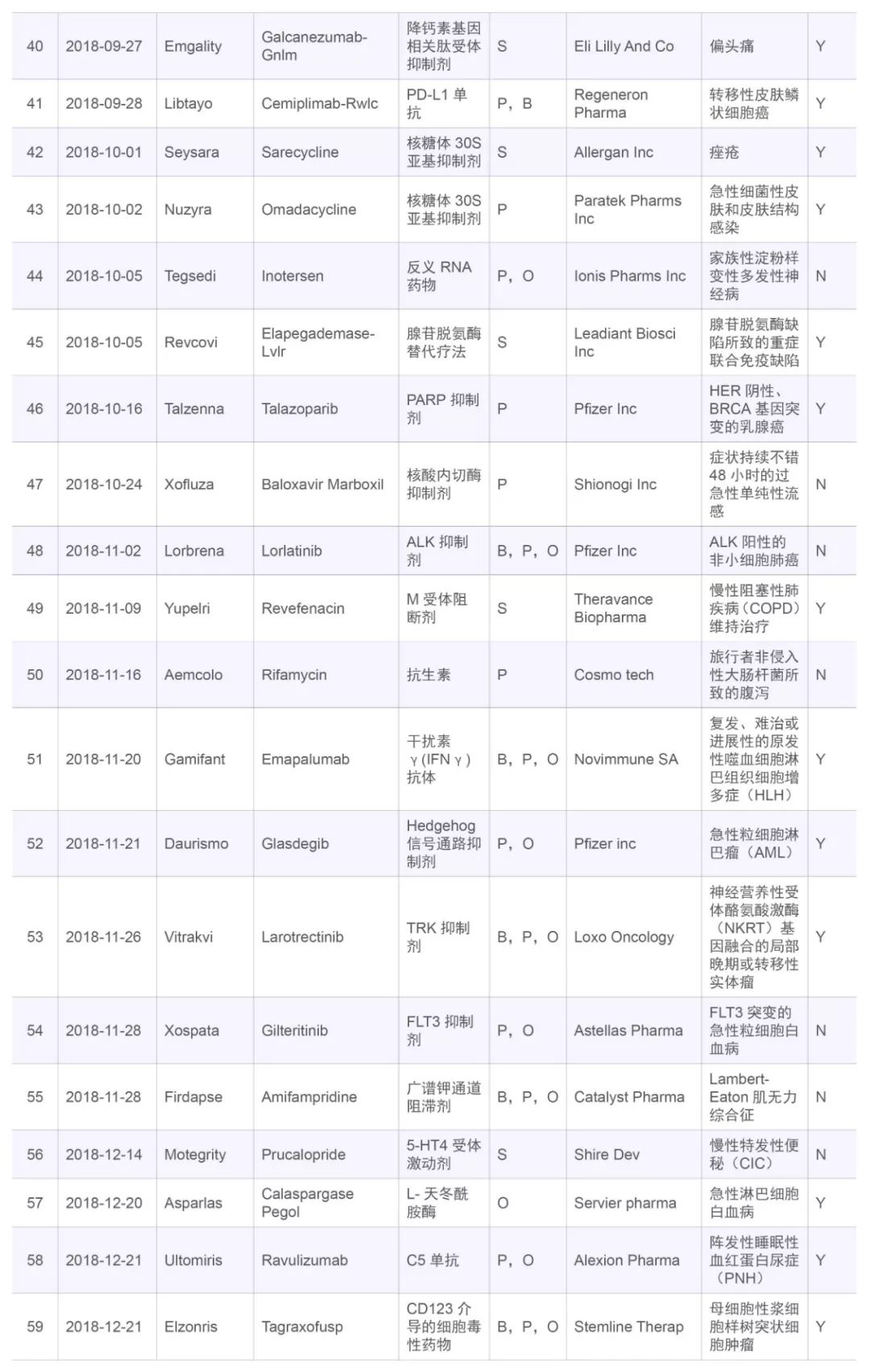
表四,2018年FDA批准的新分子实体
Table 4,FDA new drug approvals in 2018
文献参考:略










