TCGA是目前最大的癌症基因信息数据库,通过TCGA不仅可以验证你的实验结果,还可以寻找新的研究方向,围绕TCGA已经有很多重量级文章出现,对它的使用愈发重要。
目前使用的方法有两种,一是利用一些在线工具进行检索和查询,例如cBioPortal(http://www.cbioportal.org/public-portal/cgds_r.jsp)。二是下载肿瘤的三级数据,自行进行分析。
在使用这两种方法中,许多小伙伴会遇到不少问题。
对于第一种,由于在线工具大都是国外的网站,经常会遇到网速慢,网页打不开等问题,其次一些图表是由网页自行生成的,无法统一成你需要发表文章的格式,往往还需再次修改加工,所需时间不少。
对于第二种,则需要一定的生信基础和对数据库的了解才能顺利实现分析,对于大多数医生或实验研究者来说只关注自己感兴趣的数据或者只想获得一张验证或补充自己实验结果的图表而已,那么学习整个过程无疑将是费时费力的。
这期生物医学大数据解读和分析番外篇就以解决这个问题为例子,分享一个好用的R包工具,虽然涉及到一些R语言代码,但只需进行的复制黏贴代码等简单操作,就能分分钟搞定这个难题。
可点击这里,相信学习过WGCNA包的不会陌生
按一段段代码实现功能,复制黏贴到Rstudio即可,注意事项都以截图说明。
install.packages("devtools")
install.packages("ggplot2")
devtools::install_github("mariodeng/FirebrowseR")
require(FirebrowseR)
点击Rstudio中出现的cohorts,出现左上角表格,记住所需要查询的肿瘤代号,输入到红色标注代码,表示选择查询的肿瘤类型。这里以胰腺癌作为例子。
cohorts = Metadata.Cohorts(format = "csv")
cancer.Type = cohorts[grep("Pancreatic adenocarcinoma", cohorts$description, ignore.case = T), 1]
print(cancer.Type)
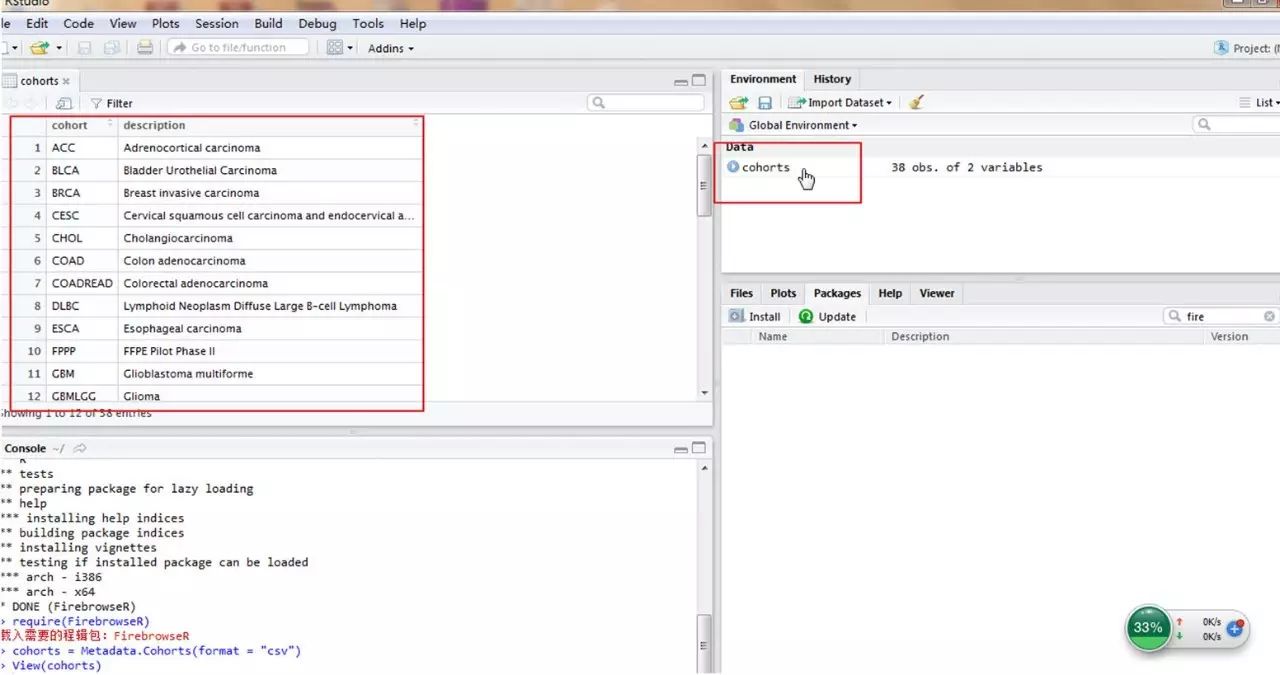
在Rstudio中点击paad.Pats,出现左上角表格,记录了所有胰腺癌的临床信息。
all.Received = F
page.Counter = 1
page.size = 150
paad.Pats = list()
while(all.Received == F){
paad.Pats[[page.Counter]] = Samples.Clinical(format = "csv",
cohort = cancer.Type,
page_size = page.size,
page = page.Counter)
if(page.Counter > 1)
colnames(paad.Pats[[page.Counter]]) = colnames(paad.Pats[[page.Counter-1]])
if(nrow(paad.Pats[[page.Counter]]) < page.size){
all.Received = T
} else{
page.Counter = page.Counter + 1
}}
paad.Pats = do.call(rbind, paad.Pats)
dim(paad.Pats)
蓝色替换成需要查询的基因,允许单个或多个基因。注意红色标记对应为相应的肿瘤代号。
diff.Exp.Genes = c("ESR1", "GATA3", "XBP1", "FOXA1", "ERBB2", "GRB7", "EGFR","FOXC1", "MYC")
all.Found = F
page.Counter = 1
mRNA.Exp = list()
page.Size = 2000 # using a bigger page size is faster
while(all.Found == F){
mRNA.Exp[[page.Counter]] = Samples.mRNASeq(format = "csv",
gene = diff.Exp.Genes,
cohort = "PAAD",
tcga_participant_barcode =
paad.Pats$tcga_participant_barcode,
page_size = page.Size,
page = page.Counter)
if(nrow(mRNA.Exp[[page.Counter]]) < page.Size)
all.Found = T
else
page.Counter = page.Counter + 1
}
mRNA.Exp = do.call(rbind, mRNA.Exp)
dim(mRNA.Exp)
mRNA.Exp = mRNA.Exp[which(mRNA.Exp$sample_type %in% c("NT", "TP")), ]
mRNA.Exp$expression_log2=as.numeric(mRNA.Exp$expression_log2)
红色标记表示y轴取值范围,可以自己调整设定。z.score代表各个基因间表达值标准化处理,一般用于多个基因放在一张图里,不仅可以比较癌和癌旁差异,还能比较各个基因间的表达差异。其中NT为癌旁, TP为癌。
library(ggplot2)
p = ggplot(mRNA.Exp, aes(factor(gene), z.score))
p +
geom_boxplot(aes(fill = factor(sample_type))) +
scale_y_continuous(limits = c(-2, 14)) +
scale_fill_discrete(name = "Tissue")

红色标记表示y轴取值范围,可以自己调整设定。expression_log2表示基因表达值的对数值,一般用于单个基因比较癌和癌旁的差异趋势。
library(ggplot2)
p = ggplot(mRNA.Exp, aes(factor(gene), expression_log2))
p +
geom_boxplot(aes(fill = factor(sample_type))) +
scale_y_continuous(limits = c(0, 8)) +
scale_fill_discrete(name = "Tissue")

在Rstudio中另存为pdf。
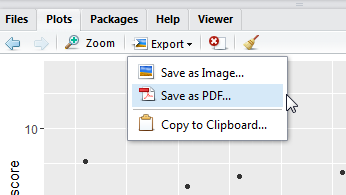
保存基因表达数据到桌面excel文件。
write.table(mRNA.Exp,file="C:/Users/Administrator/Desktop/mRNA.xls",sep="\t",quote=F)
这期番外篇讲解了如何使用TCGA数据快速做出现成的高质量图表,当然如果要修改图的格式可以学习下R作图,或者利用原始表达数据导入其他软件进行统计作图,例如GraphPad。
下期将继续根据还原文献讲解cytoscape在构建生物网络中的应用。
点公众号菜单里科研攻略-数据挖掘,查看往期系列~

赵忻艺,FS数据挖掘主编,将大数据应用于医学科研,主要包括临床医学数据的挖掘、收集、整理和利用(标准化和科学化的数据库),医学分子大数据的整理、利用及研究(基因、蛋白及代谢)。特别针对肿瘤个体化的基因测序和数据快速处理,寻找个体化的分子标志物、药物靶标和治疗方案。目前,已建立浙大大数据挖掘团队,旨在降低研究者学习大数据的门槛,推动大数据共享与研究协作,发表更高质量的研究成果,为科研决策提供精准的预测和实验证据。










