上一期向大家介绍了一篇SCI论文,
是看它如何利用已发表过的数据,通过再次提出新的问题,并回答问题
。本期向大家推荐的一篇SCI文章[
1
],
看它是如何利用文献中发表的结果,提出新的问题、解决问题。
与上一篇相似,本篇文章也是想探索吸烟是如何导致疾病发生的。
这里先给一个吸烟有害健康的
Introduction
,可能有点长,不过很有用。。。
总所周知,吸烟是一种常见的、有害的行为,能够导致一系列癌症的产生[
2
]。目前,三分之一的癌症可归因于吸烟,特别是肺癌、口腔癌、胰腺癌、食道癌、膀胱癌、喉癌和肾癌,
最为致命的癌症是肺癌和胰腺癌。
吸烟能够增加5到10倍肺癌发生的风险;在发达国家,吸烟导致的肺癌占总数的80% [
3
]。在妇女中,吸烟行为也导致的
宫颈癌
与卵巢癌的发生风险增加。相比于不吸烟者,吸烟者患
宫颈癌
的风险提高了4.4倍,而患卵巢癌的风险提高了2.8倍。
根据WHO报道
[
4
]
世界范围内每年由于吸烟导致的死亡人数估计达到了600万,其中主要原因是由于吸烟导致癌症产生的死亡。
到目前为止,许多的遗传关联分析研究已经揭示一定数量的遗传突变位点参与吸烟相关的癌症发生中[
5-7
]。
但遗憾的是,目前基于遗传突变的证据还远不能解释吸烟是如何诱导癌症产生的分子机制。
鉴于遗传突变研究现在正处于瓶颈状况,许多的科研工作者将他们的工作重心转移到与吸烟相关的
DNA
甲基化(
DNA methylation
)
上,希望通过表观遗传修饰这种分子机制来解释吸烟导致癌症的机理。
DNA
甲基化是一个可反转的遗传修饰,是最早发现的遗传修饰途径之一。大量研究证据表明,
DNA甲基化能够导致DNA
构象
、DNA稳定性、DNA与蛋白质之间的交互作用方式,以及染色质的结构发生改变,从而能够控制基因表达[
8
]、调节可变剪切[
9
],基因组的完整性[
10
]等等。
据所知,目前仍
没有一个系统全面的研究
来探索这些与吸烟相关的异常DNA甲基化位点是否参与癌症发生。
在本研究中,作者们
提出的假设
是这些已发现的与吸烟相关的异常DNA甲基化位点很可能富集在与癌症相关的基因和生物学通路上,而这些富集基因和通路在吸烟相关癌症的发生中发挥重要作用。
所以,本项研究的
主要目标
是系统全面的富集分析这些与吸烟相关的DNA甲基化位点发现重要基因和通路,并利用
TCGA
数据库
中肺癌数据对其中重要的基因进行验证。
接下来向大家展示一下,作者是如何得到他们所需要的数据的。。。
为了尽可能的找到所有关于吸烟和DNA甲基化关联的研究,作者们从PubMed数据库内总共检索到了1,447个已发表的论文(检索截止时间是2015年6月13日),
具体流程详见图
1
。所使用的关键词包括:
“Smoking”
、
“Smoke”, “Tobacco”
、
“Nicotine”
、
“Cigarette”
和
“Methylation”
。并对所有检索到的文献摘要进行阅读来
寻找到可能符合入选条件的研究
。另外,还
人工的查询
了所有合格研究的参考文献以便找到其它未被PubMed数据库索引的研究。
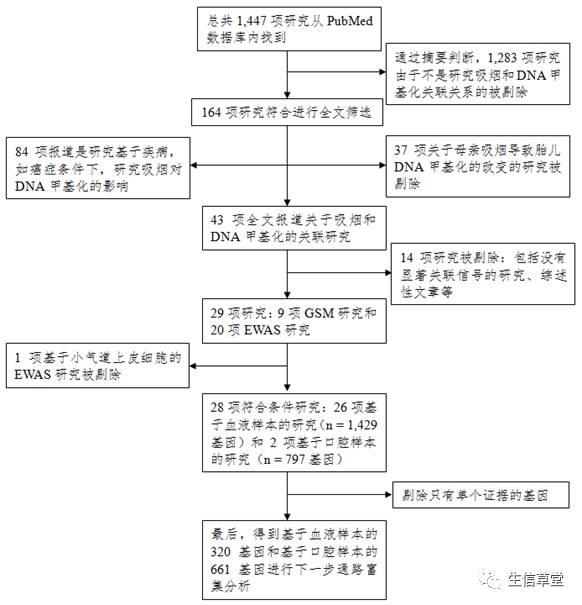
图
1
数据收集的流程图
收集好了材料,还要设定严格的入选标准,才能保证后面结果的可靠性。。。
为了尽可能的消除或减小假阳性结果的影响,设定严格入选标准,
这里只选择带有与吸烟显著关联的甲基化研究
。
一旦某项研究达到了入选标准,作者会仔细阅读该研究的全文以确保其结论与内容相符。
通过严格系统地筛选,总共有
28
项甲基化关联研究符合入选条件,其中有
26
项研究是基于血液样本(
N = 17,675
)和
2
项研究是基于口腔组织样本(
N = 1,002
)
。
同时,对于来自血液和口腔样本的两组基因进一步进行筛选以提高研究结果的可靠性。
只有基因含有两个或者以上的证据,也即是一个基因上含有两个或者以上的显著差异甲基化位点,或者一个差异甲基化位点被独立验证,才会被纳入下一步分析
。
通过设定严格的入选标准,有320个吸烟相关的差异甲基化基因从血液样本中找到。有667个吸烟相关的差异甲基化基因是来自口腔组织。
注意了:这里有个问题是,为什么基于
17,675
个血液样本只发现了
320
个基因,而基于
1,002
个口腔组织样本却发现了
667
个基因?
因为这里入选标准都是一样严格的,所以只能说明基于口腔组织的基因仍有很多假阳性基因存在。所以作者选择了用320个来自血液样本的基因作为发现阶段分析,而口腔组织的基因只是用于验证。
这一点的讨论很重要,应该在文章的
limitation
部分做讨论。不然,
reviewers
一定会提问的哦。
得到可靠的数据后,就要进行有效地数据分析啦。。
。
首先,利用来自血液样本的甲基化基因进行通路分析以发现与吸烟相关的生物学通路。然后,利用同样的方法,对来自口腔样本的基因进行通路富集分析,以验证基于血液样本发现的通路。
这里用到的通路分析软件有三个呢。。。
为了从甲基化修饰的角度全面理解吸烟对癌症产生的影响,作者利用三种生物信息学软件,包括Ingenuity Pathway Analysis (IPA;
http://www.ingenuity.com/
)[
11
]、EnrichNet (
http://www.enrichnet.org/
) 和 Genetrail (
http://genetrail.bioinf.uni-sb.de
)[
12
] 权威软件,对从血液和口腔样本中收集到的两组基因进行生物通路富集分析。
这里也要注意:尽管该研究使用了三种基于不同数据库的生物信息学软件进行信号通路富集分析,但是主要结果是由
IPA
软件产生。
这一点一定要讨论或者说明一下,不然
reviewers
会提问,如利用三个不同软件,如何考虑权衡它们之间的结果,有没有重复结果存在,是否有
Cherry-picking
的可能,等等。
在发现阶段,基于血液样本,作者利用这320个差异甲基化基因进行的通路富集分析。总共找到了90条显著富集的生物学通路(FDR < 0.05),
其中有
57
条通路是已经报道和癌症发生相关
。
例如,最显著的生物通路MSP-RON信号通路 (FDR = 2.2 × 10
-4
; 详见表1) 已经被报道参与巨噬细胞应对炎症刺激的活性调节,与上皮细胞和白细胞致癌作用相关
[
13
]
。
表
1
血液样本差异甲基化基因富集的吸烟相关的癌症通路
(FDR < 0.01)
|
富集通路
|
基因数目
|
P
值
|
FDR
值
|
|
MSP-RON 信号通路
|
8
|
6.17 × 10
- 07
|
0.00022
|
|
RAR activation信号通路
|
14
|
2.04 × 10
- 06
|
0.00037
|
|
Rac信号通路
|
10
|
6.17 × 10
- 06
|
0.00071
|
|
Actin cytoskeleton信号通路
|
14
|
7.94 × 10
- 06
|
0.00071
|
|
Aryl hydrocarbon receptor信号通路
|
11
|
1.15 × 10
- 05
|
0.00083
|
|
Rho family GTPases信号通路
|
14
|
2.51 × 10
- 05
|
0.0015
|
|
AMPK信号通路
|
12
|
2.951 × 10
- 05
|
0.0016
|
|
Renin-angiotensin信号通路
|
9
|
6.03 × 10
- 05
|
0.0028
|
|
Molecular mechanisms of cancer信号通路
|
17
|
7.41 × 10
- 05
|
0.0030
|
|
CXCR4信号通路
|
10
|
0.00017
|
0.0058
|
|
ERK/MAPK信号通路
|
11
|
0.00021
|
0.0058
|
|
HER-2 signaling in breast cancer信号通路
|
7
|
0.00021
|
0.0058
|
|
Thrombin信号通路
|
11
|
0.00022
|
0.0058
|
|
HGF信号通路
|
8
|
0.00027
|
0.0060
|
|
Relaxin信号通路
|
9
|
0.00028
|
0.0060
|
|
Role of tissue factor in cancer信号通路
|
8
|
0.00033
|
0.0063
|
|
Non-small cell lung cancer信号通路
|
6
|
0.00060
|
0.0096
|
为了
验证
基于血液样本发现的通路,作者对来自口腔样本的差异甲基化基因进行了相同的通路富集分析,找到了32条共有通路(p < 0.05)。
在这些共有通路中,有








