近年来,NGS和质谱技术的进步显著推进了癌症研究,
大型研究项目
如癌症基因组图谱(TCGA)、国际癌症基因组联盟和临床蛋白质组学肿瘤分析联盟(CPTAC)等提供了对癌症的深入理解,并发现了许多体细胞突变和癌症相关蛋白。
蛋白质互作网络对于确定体细胞突变的优先次序和预测未表征蛋白质的功能至关重要,但现有许多相互作用是在非癌症环境中发现的,其与癌症的相关性尚不明确。近期的研究试图通过分析特定癌细胞系中蛋白质的相互作用来解决这一问题,
但全面无偏倚地分析不同癌症类型中的蛋白质相互作用
仍充满挑战
。
近日,美国贝勒医学院章冰团队在
Nature Cancer
发表了题为“Mapping the functional network of human cancer through machine learning and pan-cancer proteogenomics”的文章。
研究团队基于监督机器学习方法,结合11种癌症类型中1,194个个体的大量蛋白质组学和RNA测序(RNA-seq)数据构建了一种名为
FunMap的
泛癌功能网络
。FunMap包含
10,525个蛋白质编码基因
,超越了传统的蛋白质-蛋白质相互作用图谱,不仅加深了对现有癌症驱动因素提供的理解,还能预测未知癌症相关蛋白的功能,对癌症生物学发展和指导治疗策略具有重要意义。
研究团队纳入了来自11个肿瘤队列的质谱蛋白质组学数据和RNA-seq数据,涉及1,194名患者的肿瘤样本。通过
对于每个蛋白质组学和RNA-seq数据集进行分析,研究团队发现
蛋白质组学数据比RNA-seq产生更高功能相关性
。
研究团队还探究了肿瘤数据集中mRNA和蛋白质共表达模式与基因协同功能的关系,发现虽然蛋白质和mRNA的相关性都表明基因协同功能,但
蛋白质相关性是一个更强的预测因子。
此外,研究团队利用监督机器学习并将其与蛋白质组学和RNA-seq数据集中各种预测信号进行整合,构建了一个全面的功能网络。
特征重要性分析显示,肿瘤蛋白特征贡献最大,其次是肿瘤mRNA、正常mRNA和正常蛋白。通过
进一步
筛选出
高可信度的基因对,
最终生成了一个包含10,525个基因和196,800个节点的功能关联网络——FunMap
。
与其他网络相比,
FunMap
存在大量独特功能关联,
其边缘连接的
60-74%基因是
肿瘤中显著过表达或低表达的基因,这显示出
FunMap与癌症的强关联
。此外,FunMap还具有相对较高的平均聚类系数、相对较高的密度等。总之,
F
unMap具有高功能相关性、癌症相关性和蛋白质组覆盖率。
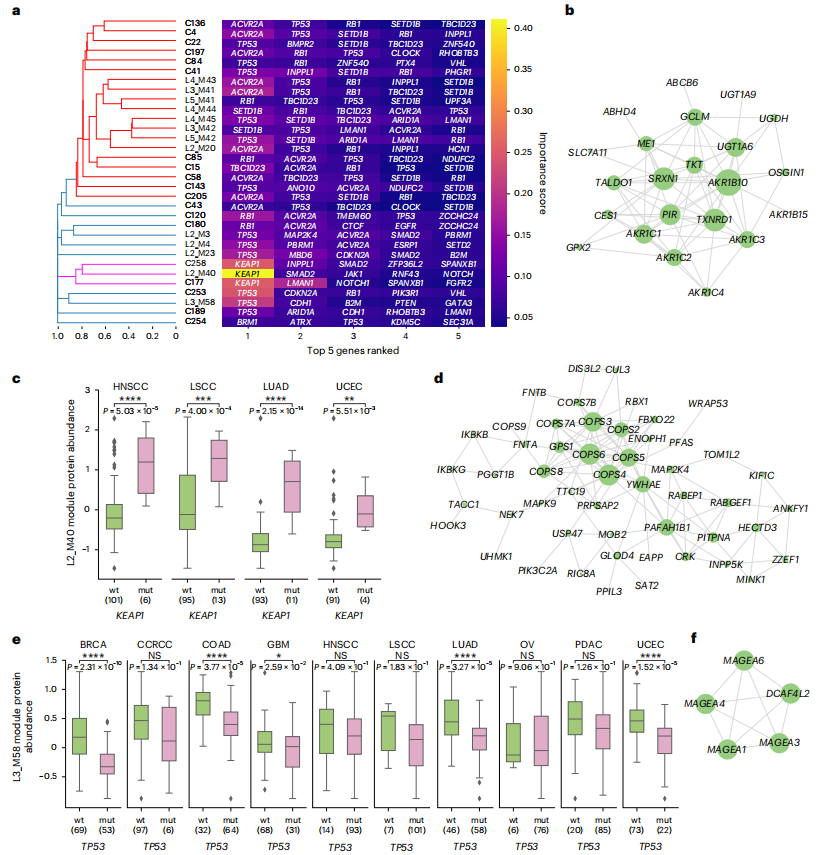
癌症蛋白质基因组学的一个主要目标是了解体细胞突变如何影响癌症蛋白质组。研究团队使用机器学习方法同时分析了FunMap中所有重要突变对单个功能模块的影响,发现
32个模块在预测丰度和实际丰度之间存在非随机相关性,这表明这些模块的突变状态与蛋白质丰度密切相关
。
在所有32个模块中,TP53是最重要的预测因子
,而其他基因特定于某些模块。总之,FunMap有效地将体细胞突变与各功能模块中的蛋白质丰度相关联,并
能识别出关键突变基因,有助于揭示未被充分研究的癌症蛋白。
基于图神经网络(GNN)的深度学习,研究团队还开发了一种正样本未标记学习算法,该算法集成了FunMap网络,并从CPTAC队列和已知的癌症基因中提取基因突变显著性评分。研究团队借助上述算法
训练了一个图注意力网络模型(GAT),用于将未标记的基因分类为癌症或非癌症基因
。
性能评估显示,
FunMap GAT模型的分类准确性优于不使用网络数据训练的随机森林分类器
,其AUROC提高了6.5%,AUPRC提高了27.8%。此外,
FunMap在识别低突变频率基因作为假定癌症基因方面也具备有效性。
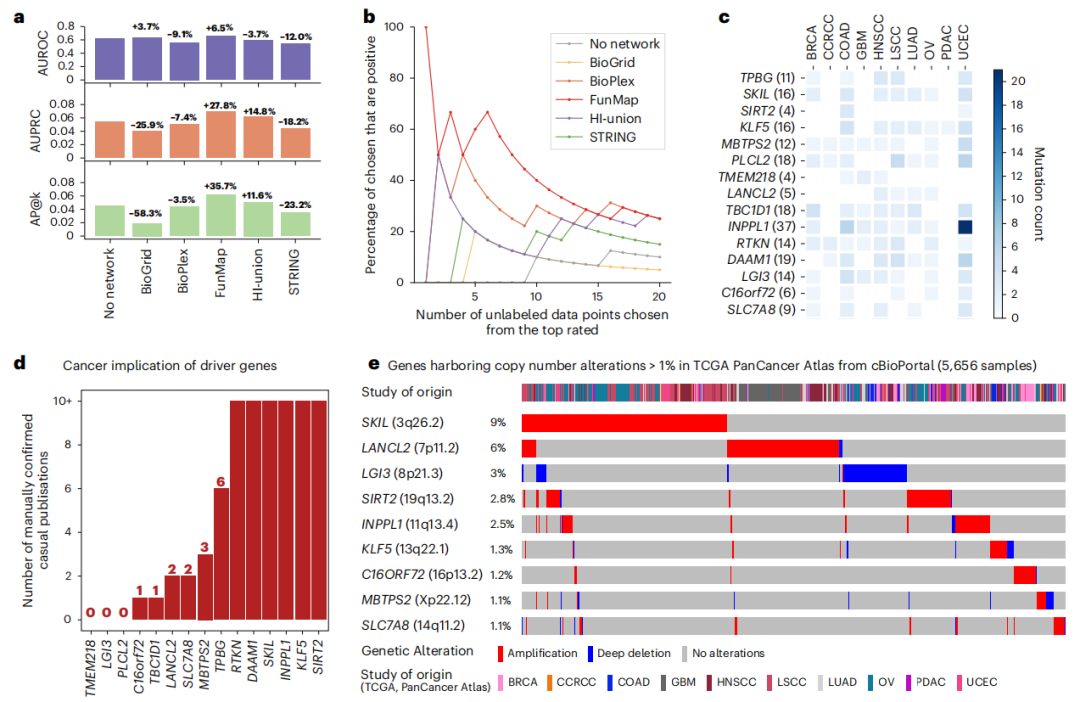
图4. 利用FunMap发现低突变频率的癌症驱动因子
FunMap
通过结合机器学习和蛋白质基因组分析的优势,提供了一个全面且无偏颇的蛋白质组学覆盖和高水平的功能相关性,可更深入地了解复杂的癌症系统,为体细胞突变和癌症相关蛋白研究提供了有价值的见解。同时,FunMap作为
一种补充方法,
丰富了蛋白质互作网络研究,扩展了功能基因组学研究的系统生物学框架。
Shi, Z., Lei, J.T., Elizarraras, J.M. et al. Mapping the functional network of human cancer through machine learning and pan-cancer proteogenomics. Nat Cancer (2024).https://www.nature.com/articles/s43018-024-00869-z
·END·










