McscanX是进行基因水平共线性分析的较为常用的软件,使用此软件可以得到共线性区块(block),利用得到的文件可以进行共线性作图。
一.输入文件
1.Blast文件。BLAST为你要分析物种基因的蛋白质文件或者CDS文件与参考物种的蛋白质或者CDS文件的BLAST比对结果文件,格式一般为采用BLAST的m8或者BLAST+的 -outfmt 6格式。此处以BLAST+为例阐述.
1)blast建库:
$ makeblastdb -in refpep.fa -dbtype prot -out refpep.db |
2)blast比对:
$ blastp -query yourpep.fa -db refpep.db -out all.blast -evalue 1e-10 -num_threads 4 -outfmt 6 -num_alignments 5 |
-evalue 1e-10
-num_alignments 5是取最好的5个比对结果,
-outfmt 6是输出格式为tab分隔的比对结果。
3)示例结果如下:

2.Gff文件。Gff文件为上述用于BLAST的基因在染色体上的位置信息文件,包括你要分析物种的和参考的基因在染色体上的位置信息。具体格式为:

其中sp#:sp代表物种名字,#代表相应的染色体或者scaffold名字;其中加入sp目的是主要为了区分参考物种序列名字和你要分析物种序列名字.
你要分析物种的和参考的信息最终应合并到一个文件中。我们可以将文件命名为all.gff。
3.注意事项.
1)blast和gff文件命名规则为*.gff和.blast,即后缀名不变,前缀名字必须一致!
2)blast和gff文件里面的基因ID必须名字必须一致!
二.运行命令
参数介绍
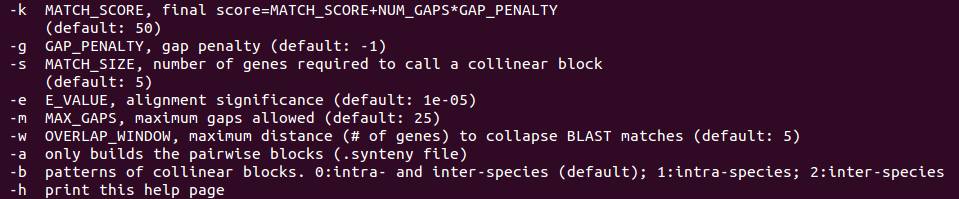
主要参数介绍:
-s:要求一个共线性区块应具有的最少基因数目
-m:一个block中允许的最大空位数目
-a为选择物种间的共线性还是物种内部共线性
2.运行命令
$MCScanX/MCScanX all
三.结果介绍
生成一个文本文件和网页格式文件。
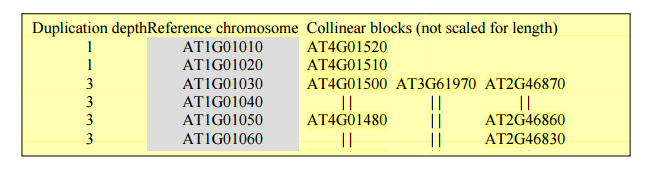
1. 文本文件给出了每一个Block由那些基因组成,大伙直接查看或者处理这些数据做Circos图。

2. 网页格式文件,可能更加直观,可以清楚的看到每个block内基因的对应关系。并且能看到具体的某个基因参与共线性block的个数
四.共线性图绘制
可以画四种图如下:
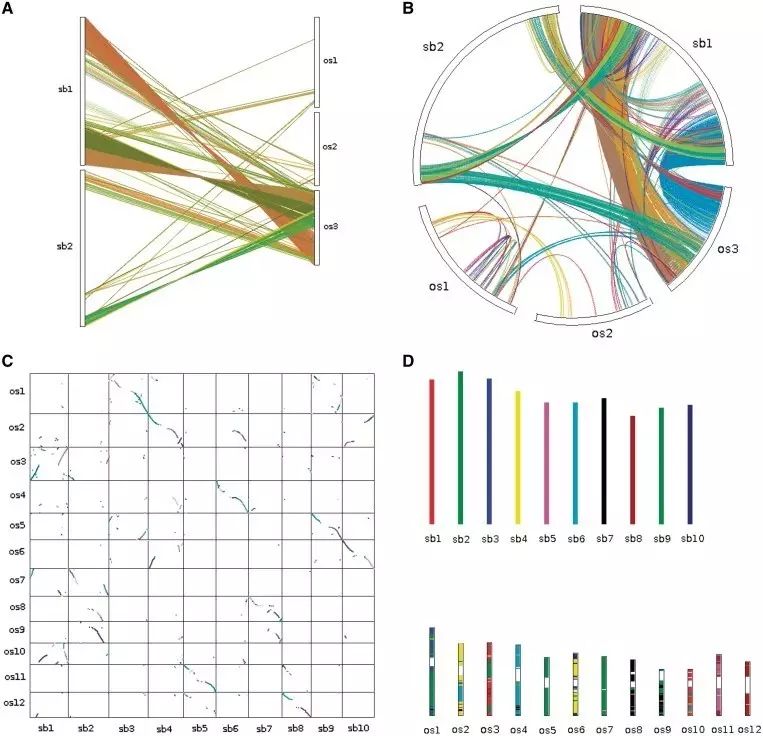
(A) dual synteny plot, (B) circle plot, (C) dot plot and (D) bar plot
1.dual_synteny_plotter.java
$ java dot_plotter -g gff_file -s collinearity_file -c control_file -o output_PNG_file |
其中control文件格式如下:

2. circle_plotter.java画圆圈图
$ java circle_plotter -g gff_file -s collinearity_file -c control_file -o output_PNG_file |
其中control文件格式如下:
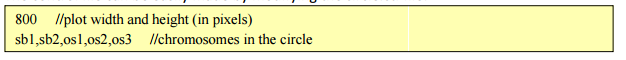
3. dot_plotter.java画dotplot图
$ java dot_plotter -g gff_file -s collinearity_file -c control_file -o output_PNG_file
其中control文件格式如下:

4. bar_plotter.java画柱状图
$ java bar_plotter -g gff_file -s collinearity_file -c control_file -o output_PNG_file
其中control文件格式如下:

欢迎关注生信人











