这是一个关于高通量基因组学研究相关基础知识的系列内容,旨在为初步涉入基因组学研究的同学查缺补漏,答疑解惑。如果前期对基本概念都没理解通透,基础没打好,后期会走弯路的。本系列内容涉及基因组学、高通量测序相关基本概念,基因组分析中常见问题等,每期
5-10
个
FAQ
,希望对大家有用。大家有其他相关的问题,可以在后台留言,我们会尽力在下棋为您解答
基因组组装常见问题:
Q1
:在有杂菌污染的情况下,为什么得不到好的组装结果呢?
A1:
如果是近源物种,二者的
GC
含量相似,具有大量的同源序列;非近源物种
DNA
污染,虽然可以通过
GC-depth
将非近源物种的一些序列找出并剔除,但是其
DNA
中会有不同程度的相似性序列;高度相似序列会对组装软件产生干扰,而软件为保证组装的准确性,只能将可疑的部分切断成不同的碎片序列
,
从而形成很多个
contig
,从而很难形成一个完整的环,导致组装结果不好。
基因组注释知识锦集:
Q1
:什么是基因组注释?
A1
:基因组注释
(Genomeannotation)
是利用生物信息学方法和工具
,
对基因组所有基因的生物学功能进行高通量注释
,
是当前功能基因组学研究的一个热点。基因组注释的研究内容包括基因识别和基因功能注释两个方面。基因识别的核心是确定全基因组序列中所有基因的确切位置。
Q2
:具体讲解一下
KEGG
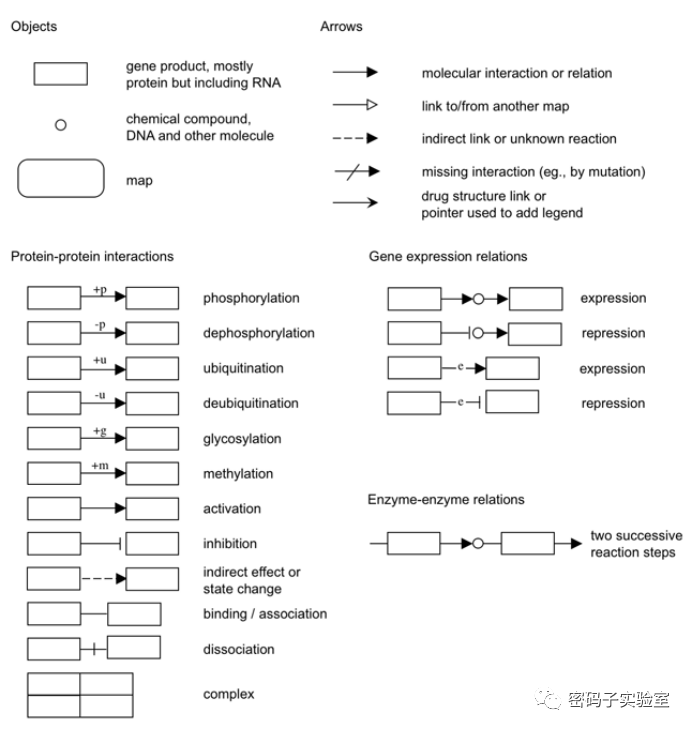
代谢通路图,图中的箭头,虚线箭头分别代表什么?
A2:在KEGG代谢通路图中,实线箭头有以下几种含义:(
1
)一步生化反应;(
2
)正向调控;(
3
)分子相互作用
而虚线箭头的意义:(1)与其它
pathway
的连接(表示间接反应);(
2
)不确定的生化反应。
具体见下图:
Q
3
:
KEGG
注释里面的
KO
和
ko
分别代表什么?
A
3
:
KO
是基因的编号,代表某一类同源基因,同源基因在不同的物种/基因组中会有不同的基因名,但它们的
KO
编号是一致的;
ko
是通路的编号,代表某一个通路。
Q4
:将某一基因的
KO
号输入
KEGG
pathway
,为什么没有找到与之对应的通路呢?
A4
:
KEGG
库中注释到的基因,有一部分是参加代谢网络的或者有代谢通路图,可以在
KEGG
的
pathway
数据库中找到,但是有一部分基因是不参加代谢通路网络的,或者是
KEGG
的
pathway
数据库现有的代谢通路图中没有该基因参与的代谢通路图,这部分基因只能在
KEGG
的
gene
库中找到,不能在
pathway
数据库中找到。
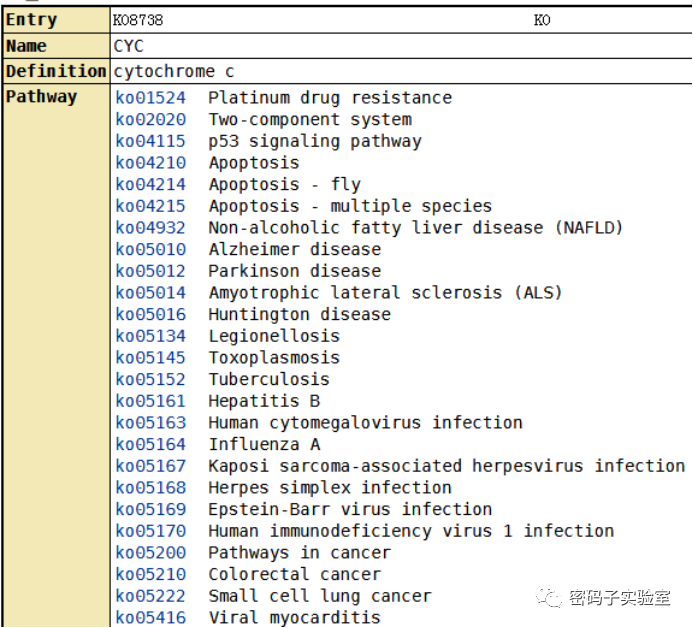
Q5:
为什么原核生物基因组会注释到真核生物的代谢通路?
A5
:这里有
2
种可能:一是该基因在原核物种的数据库中未能找到很好的匹配结果,而在真核生物中的某个被归类到真核代谢通路的基因与该基因达到最佳匹配,因此被注释到的基因也就展示了真核代谢通路;二是展示代谢通路时,它的逻辑并不仅仅展示该基因对应的代谢通路,而是展示该基因注释到的
KO
对应的所有代谢通路。比如细胞色素
C
(
K08738
),这类同源基因在细菌中可以参与双组分调控系统,而在人体内与耐药性、疾病、癌症等多种代谢途径有关。因此,即使实际情况下某个细菌的
K08738
基因本身只与双组分调控系统有关,最终还是会显示一些与细菌代谢无关的通路。总之,这种情况无需过分纠结,根据实际情况进行判断即可。
Q6:
在
pathway
数据库中,这个下拉菜单分别代表什么?
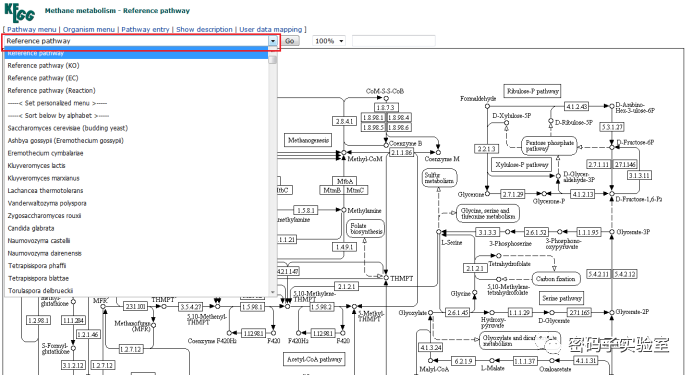
A6
:
Reference pathway
表示没有特别标注的代谢通路。
Reference pathway
(
KO
)是把
KO
数据库中所有的数据填充在代谢通路中;
Reference pathway
(
EC
)是把酶数据库所有的数据填充在代谢通路中;
Reference pathway
(
Reaction
)是把反应数据库所有的数据填充在代谢通路中;下面就是把各个物种所包含的
KO
注释填充在代谢通路中。
Q7
:
KEGG
注释能否精确到具体某个物种?
A7
:线上注释在选择数据库的时候可以只选择某个物种进行注释。
Q8
:通过实验已经证明菌株中具有某种功能的基因,而在注释结果里面没有,是什么原因?
A8
:首先,虽然我们通常认为具有相似序列的蛋白具有相似的功能,并主要通过序列相似性来进行基因注释。但实际情况下,这种假设并不是绝对的,即序列相似性并不完全等价于功能相似性。有的基因会由于一个碱基的
SNP
完全丧失或改变原有的功能,也有的基因在很低的序列相似性情况下也具备相同的功能。因此,基于序列相似性的基因注释是无法注释到这些基因的。
除此之外,基因没有被注释到也有可能与基因组装、基因预测有关,例如含有该基因的
DNA
片段由于组装错误丢失了,或者该基因在预测过程中由于预测软件的关系没有被预测是基因,这些原因都有可能造成注释的缺失。
基因组重测序知识锦集:
Q1
:什么是重测序?
A1:
即对已知基因组序列的物种进行不同个体的基因组测序,是一种有参分析,目的在于寻找不同个体或者群体的基因组变异差异,通过序列比对,找到单核苷酸多态性位点(
SNP
),插入
/
缺失位点(
InDel
)、结构变异位点(
SV
,
Structure Variation
)位点和拷贝数变异位点(
CNV
,
Copy Number Variation)
。
Q2
:什么是
INDEL (
基因组小片段插入)?
A2
:
INDEL
指的是在基因组的某个位置上所发生的小片段序列的插入或者删除,其长度通常在
50bp
以下。
Q3
:什么是
copy number variation
(
CNV
):基因组拷贝数变异?
A3
:基因组拷贝数变异是基因组变异的一种形式,通常使基因组中大片段的
DNA
形成非正常的拷贝数量。例如人类正常染色体拷贝数是
2
,有些染色体区域拷贝数变成
1
或
3
,这样,该区域发生拷贝数缺失或增加,位于该区域内的基因表达量也会受到影响。
比较基因组学:
Q1
:什么是比较基因组学?
A1
:比较基因组学
(Comparative Genomics)
是基于基因组图谱和测序基础上,对已知的基因和基因组结构进行比较,来了解基因的功能、表达机理和物种进化的学科。
扩增子测序
与菌种鉴定知识与锦集:
Q1
:为何有些细菌能够鉴定到种水平,有些只能鉴定到属水平?
A1
:目前基于
16S rRNA
进行种属鉴定,到底能够到什么程度,主要依赖于
NT
数据库的已公开序列,如果某个物种研究比较多,可能会有鉴定到种甚至鉴定到亚种、变种的菌株被公开。反之,如果某个物种研究较少,特别是还没有任何全基因组被测序的物种,往往存在鉴定不细致、不准确的情况,很多科研者只鉴定到属就投递了。
Q2
:为何用
16S rRNA
比对会比对出不同的种属?
A2
:由于历史的原因,一些物种前期可能被划归到某个种或者某个属,随着研究的深入,又被重新划归到其他的种属里面,故而会出现同样的序列被鉴定成不同的种属的情况。研究者要根据现在的研究方向和研究进展做以判断。
Q3
:有哪些方法可以鉴定物种种属?
A4
:最常用的方法是
16S rRNA
,另外,还有很多学者使用
MLSA
的方法基于更多的保守基因构建进化树进行物种种属判定。目前比较流行的、更加准确的方法是基于全基因组单拷贝基因的物种进化树和基于全基因组序列的
ANI
分析。
Q5
:根据
16S rRNA
如何判断样品污染?
A5
:观看测序峰图,常见的污染类型表现为高度的重叠峰或者轻微的双峰。分为不同种属样品污染,少量近源物种之间的污染,以及同种不同株之间的污染。
猜你喜欢
微生物基因组公开课回放
1.
细菌基因组测序方式:重测序、扫描图、完成图、转录组如何选择
2.
细菌基因组研究思路和案例分享
3.
常见的比较基因组分析有哪些
5.PC电脑上如何绘制高水平的基因组圈图?
6.如何使用BRIG绘制比较基因组圈图?
7.如何通过BLAST软件进行比较基因组分析?
8.BRIG高级使用与常见问题?
脚本大放送
火山图
Venn图
upset图
fasta序列提取
科研绘图
AI使用
AI绘图-常见问题汇总










