天子呼来不上船,
自称臣是菜鸟团。
在这里,和国际同行一起学习单细胞数据分析。
关注Seurat的github动态的朋友不难发现,其实许许多多问题是反复出现的,如本期的封面故事:
Resolution parameter in Seurat’s FindClusters function for larger cell numbers
即,在做细胞分群的时候如何确定分群数据量?在这之前我们先来看个有趣的问答吧。
Error using WhichCells/subset in a for loop
讨论池:
https://github.com/satijalab/seurat/issues/1053
https://github.com/satijalab/seurat/issues/1839
当我们需要循环地对某个基因列表用
WhichCells/subset
做条件筛选的时候:
genes v for (i in v){
cells 0)
print(length(cells))
}
我们发现:
Error in FetchData(object = object, vars = expr.char[vars.use], cells = cells, : None of the requested variables were found:
而单独使用基因名是可以的:
WhichCells(seurat_object, expression = KIF22 > 0)
为什么呢?为什么基因名作为循环变量就不行了呢?我们发现循环的时候基因名是一个字符串,而单独写的时候并不是字符串(没加引号),GitHub的回答是:
Much like R’s
subset
function,
subset.Seurat
is designed for interactive use only. While we currently don’t offer a programmatic way to subset Seurat objects based on feature expression, this can be accomplished relatively easily using which and
FetchData
:
var1 expr object[, which(x = expr > low & expr < high)]
所以在我们设计循环体的时候,不能直接用
WhichCells/subset
,可以用上面的方式:
library(Seurat)
gene_cells = list()
items for (gene1 in items)
{
if (gene1 %in% rownames(pbmc_small))
{
expr gene_cells[[gene1]] = 0.5)]
}
}
gene_cell
$LGALS3
An object of class Seurat
230 features across 24 samples within 1 assay
Active assay: RNA (230 features)
2 dimensional reductions calculated: pca, tsne
$S100A11
An object of class Seurat
230 features across 45 samples within 1 assay
Active assay: RNA (230 features)
2 dimensional reductions calculated: pca, tsne
关于Seurat的subset还有一个诡异的地方:
pbmc_small[VariableFeatures(object = pbmc_small), ]
pbmc_small[, 1:10]
subset(x = pbmc_small, subset = MS4A1 > 4)
subset(x = pbmc_small, subset = `DLGAP1-AS1` > 2)
subset(x = pbmc_small, idents = '0', invert = TRUE)
subset(x = pbmc_small, subset = MS4A1 > 3, slot = 'counts')
subset(x = pbmc_small, features = VariableFeatures(object = pbmc_small))
当基因名中有-的时候,需要用反单引号引起来才能行。
下面是封面故事
我的细胞到底分多少个群是合适的?这是一个广泛而经典问题。就单细胞技术而言,我们常说每个细胞都是不同的,也就是说你总可以分到最细以单细胞为单位,但是这样就失去高通量的意义了。在低通量下,我们可以着眼于单个细胞,现在成千上万的细胞,一个一个看是不切实际的。那么,我的细胞到底分多少个群是合适的?
Resolution parameter in Seurat’s FindClusters function for larger cell numbers
讨论池:
bioinformatics : https://bioinformatics.stackexchange.com/questions/4297/resolution-parameter-in-seurats-findclusters-function-for-larger-cell-numbers
github :https://github.com/satijalab/seurat/issues/1565
这个问题表现在Seurat中就是:Finding optimal cluster resolution in Seurat 3? 我们知道,不同的resolution参数会带来不同的分群结果。先看一下github上面的回答:
While Seurat doesn’t have tools for comparing cluster resolutions, there is a tool called clustree designed for this task and works on Seurat v3 objects natively. It’s available on CRAN and can be installed with a simple install.packages(‘clustree’)
clustree我们之前讲过,可以全局地查看不同分群结果:
#先执行不同resolution 下的分群
library(Seurat)
pbmc_small object = pbmc_small,
resolution = c(seq(.4,1.6,.2))
)
clustree([email protected], prefix = "RNA_snn_res.")
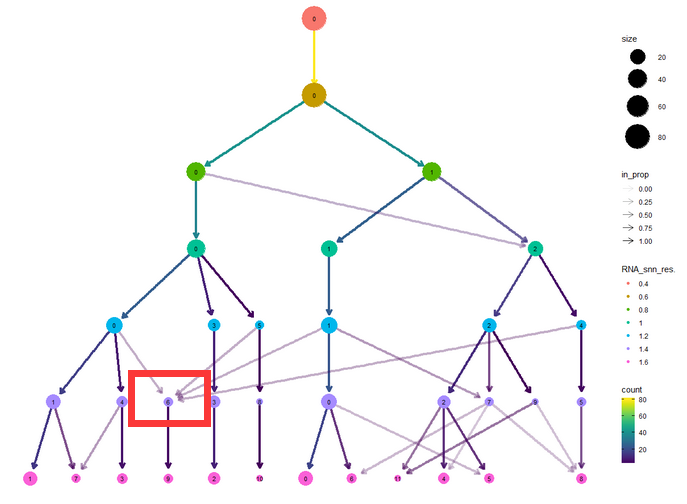
在clustree的图中我们看到不同resolution的取值情况下分群的关系。既然我们最终是以群为单位来分析的,我们肯定是希望每个群是比较纯的。如图可以看到在倒数第二层级有个亚群来自不同的分群,这有可能是:










