这周来点儿热乎的东西吧!最近要分析一批SNP数据,所以今天跟大家分享的是关于SNP建树的软件SNPhylo。之所以会选择这个软件,是因为之前在看藜麦基因组的文章时发现,这可是发在Nature正刊上的文章,这么厉害的文章都用它,那应该不会错。这不是广告,只是当你不知道用什么软件来分析你的数据时的一条捷径。看文献!!!来一睹为快:

系统进化树是一个很好的工具来推断各种生物之间的进化关系,所以系统进化树已被用于许多进化研究。基于SNP数据的系统发育树已经在重测序项目广泛应用。然而,并没有简单的方法来从重测序数据中的大量变异位点来确定系统发育树。因此,我分享一个新的流程,snphylo,构建基于SNP数据的系统发育树。使用此流程,用户可以从包含大量SNP数据的文件中构建系统树。
它有三大特点:
基于Genome wide的SNP构建进化树。传统进化树的构建大多数是是基于单拷贝基因、核糖体RNA基因、内转录间隔区序列等具有特定性质的序列。snphylo构建全基因组信息进化树,因此,它是更准确。
通过连锁不平衡来减少SNP冗余。SNPs在同一连锁不平衡区域中有大量冗余家系信息。snphylo在一个连锁不平衡区域只保留其中的一个SNP的信息。它大大减少了运行时间而不会遗漏有效信息位点。
自动化的建树流程。SNPhylo以最常见的SNP/基因型(VCF /hapmap)作为输入,只通过一条命令就可以得到一颗最大似然树!
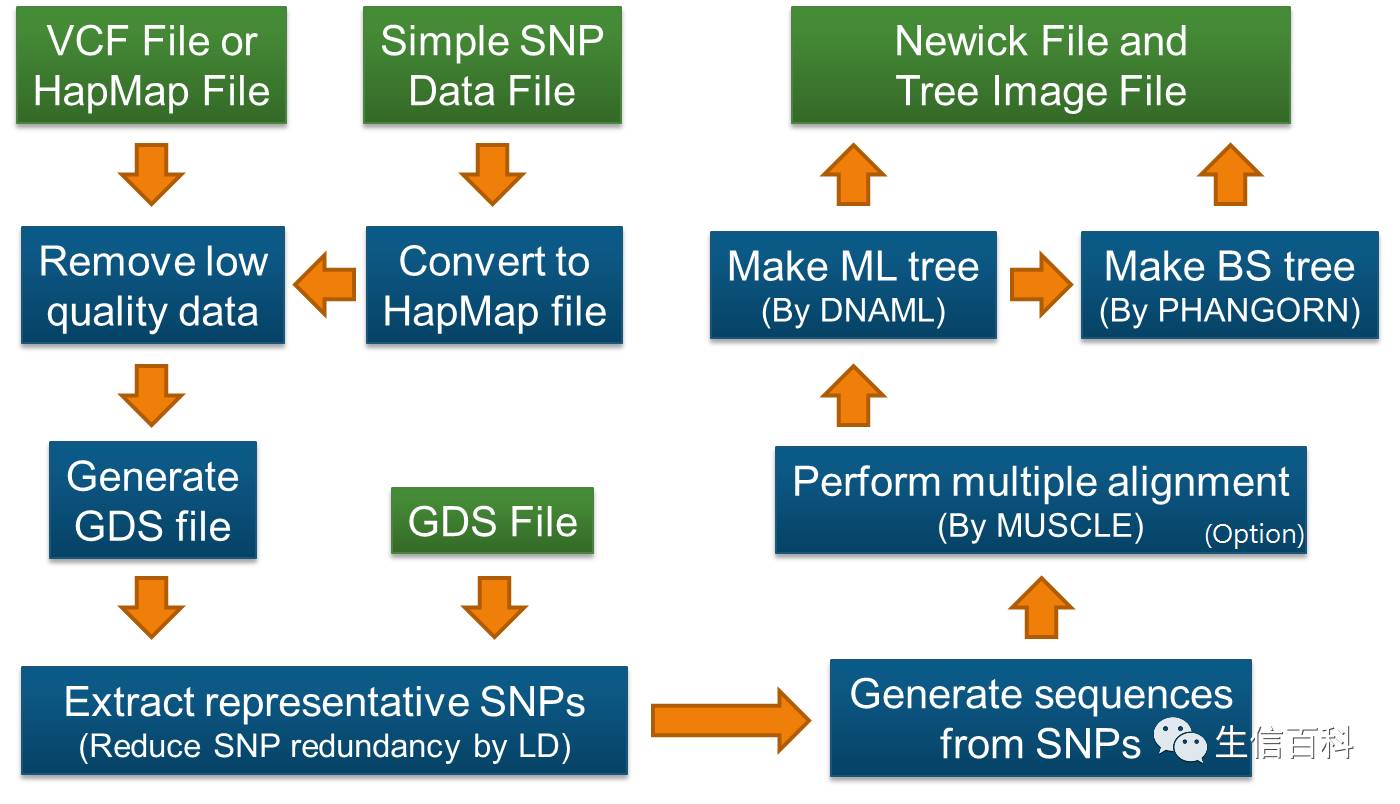
建树流程图
安装SNPhylo依赖的软件
R
R属于服务器标配
安装R包
install.packages("getopt", repos="http://cran.r-project.org")
install.packages("phangorn", repos="http://cran.r-project.org")
source("http://bioconductor.org/biocLite.R")
biocLite("gdsfmt")
biocLite("SNPRelate")
我所遇到的Error
Error1:
Error: libiconv.so.2: cannot open shared object file: No such file or directory
原因:缺少库文件解决方案:
wget http://ftp.gnu.org/pub/gnu/libiconv/libiconv-1.11.tar.gz
tar -xvzf libiconv-1.11.tar.gz
cd libiconv-1.11
./configure --prefix=/usr/local/libiconv
make
sudo make install
su root
cd /usr/local/lib
ln -s /usr/local/libiconv/lib/libiconv.so.2.4.0 libiconv.so.2
Error2:
ERROR: dependency ‘htmlwidgets’ is not available for package ‘htmlTable’
ERROR: dependency ‘htmlTable’ is not available for package ‘Hmisc’
原因:R包之间的相互依赖关系
解决方案:
install.packages("htmlwidgets", repos="http://cran.r-project.org")
install.packages("htmlTable", repos="http://cran.r-project.org")
install.packages("Hmisc", repos="http://cran.r-project.org")
如果你也遇到与我同样的Error信息,可以参照我的解决办法
Python
Python也属于服务器标配
MUSCLE
wget http://www.drive5.com/muscle/downloads3.8.31/muscle3.8.31_i86linux64.tar.gz
mkdir muscle
tar zxf muscle3.8.31i86linux64.tar.gz -C muscle
ln -s muscle3.8.31_i86linux64 muscle
echo 'PATH=$PATH:/path/to/muscle/' >> ~/.bashrc
source ~/.bashrc
Phylip
wget http://evolution.gs.washington.edu/phylip/download/phylip-3.696.tar.gz
tar zxf phylip-3.696.tar.gz
cd phylip-3.696/src
make -f Makefile.unx install
echo 'PATH=$PATH:/path/to//phylip-3.696/exe/' >> ~/.bashrc
source ~/.bashrc
SNPhylo
wget http://chibba.pgml.uga.edu/snphylo/snphylo.tar.gz
tar zxf snphylo.tar.gz
bash setup.sh
"""Version: 20141127
START TO SET UP FOR SNPHYLO!!!
The detected path of R is /path/to/R-3.2.0/bin/R. Is it correct? [Y/n] yThe detected path of python is /path/to/python. Is it correct? [Y/n] yThe detected path of muscle is /path/to/muscle/muscle. Is it correct? [Y/n] yThe detected path of dnaml is /path/to/phylip-3.696/exe/dnaml. Is it correct? [Y/n] ySNPHYLO is successfully installed!!!"""
到此安装成功!!!
查看帮助文档
./snphylo.sh -h
示例:
测试数据下载:
wget http://chibba.pgml.uga.edu/snphylo/soybean.hapmap.gz
gunzip soybean.hapmap.gz
运行
./snphylo.sh -H soybean.hapmap -P soybean -B 100 -A -b
结果解读:
soybean.bs.png:带有自展支持率的ML树

soybean.bs.tree:Newick 格式文件,带有自展支持率的ML树文件,可在FigTree中打开

soybean.fasta:输出比对后的fasta格式文件
soybean.filtered.hapmap:过滤后的数据
soybean.gds:从过滤文件生成的GDS文件
soybean.ml.png:ml树
soybean.ml.tree:Newick 格式文件
soybean.ml.txt:DNAML的输出文件(DNAML来自phylip软件包)
soybean.hapmap:hapmap格式的输入文件
soybean.id.txt:用于建树的ID列表文件(这里的ID就是vcf文件中的ID)
soybean.phylip.txt:PHYLIP格式的序列文件,可以用这个文件做其他的分析
参数解读:
./snphylo.sh -h
Determine phylogenetic tree based on SNP data with a VCF, a HapMap, a Simple SNP or a GDS file
Version: 20140701
Usage:
snphylo.sh -v VCF_file [-p Maximum_PLCS (5)] [-c Minimum_depth_of_coverage (5)]|-H HapMap_file [-p Maximum_PNSS (5)]|-s Simple_SNP_file [-p Maximum_PNSS (5)]|-d GDS_file [-l LD_threshold (0.1)] [-m MAF_threshold (0.1)] [-M Missing_rate (0.1)] [-o Outgroup_sample_name] [-P Prefix_of_output_files (snphylo.output)] [-b [-B The_number_of_bootstrap_samples (100)]] [-a The_number_of_the_last_autosome (22)] [-r] [-A] [-h]