作者 | 朱玉苹
本文经微信公共号“蝌蚪士”授权转载

肿瘤和炎症之间的相互关系在科学研究中长期存在着争议。1863 年,德国病理学家Rudolph Virchow认为肿瘤起源于慢性炎症部位,既组织损伤和随后的炎症是产生肿瘤的重要原因 [1]。现研究表明,许多肿瘤来自于受感染、慢性刺激的炎症部位,其中由炎症细胞组成的肿瘤微环境,是导致肿瘤发生的一个重要因素,即急慢性炎症的长期刺激增加了罹患肿瘤的风险,并促进了肿瘤的发生。尽管现已研究表明,细胞异常增殖并不能一定会导致肿瘤的发生,如组织受到损伤时会进行自我修复,相关部位组织细胞增殖也会增加,当修复完成或攻击介质移除后,细胞增殖和炎症反应会最终得到消退。而炎性细胞以及炎性细胞周围的正常细胞在具有丰富的活化的基质、生长因子,以及DNA 受到损伤等促进因素长期刺激下细胞增殖将持续进行,从而进一步增强肿瘤发生的风险。另外,由细菌、病毒等感染因素引起的慢性炎症也会造成人类肿肿瘤的发生。肿瘤细胞会主动选如选择素、趋化因子及其受体,促进自身的的侵袭、转移[2]。然而,炎症、先天免疫和肿瘤之间的相互关系虽然得到了更广泛地接受与认可,但是由许多分子和细胞机制介导的这种关系仍然没有理清。此外,虽然获得性免疫应答的癌症与炎症反应密切相关[3,4],但导致肿瘤细胞发生的关键机制目前仍然没有形成定论。
1 炎症与肿瘤的相互关系的研究现状
1.1炎症和肿瘤关系的起源
Peyton Rous等,最先提出肿瘤是由病毒或化学致癌物诱导体细胞改变引起的观点[5,6]。许多启动子在肿瘤的发生过程中都会诱导细胞增殖,减少并降低基因的自我修复功能。长期发炎的组织细胞的死亡和修复会导致细胞的恶性增殖的机率。正常情况下,机体遭受损伤形成炎症反应的同时,促炎症因子或者炎症因子会很快激活抗炎细胞因子信号通路,促使抗炎细胞因子产生,实现机体的自我保护能力。然而,在慢性炎症持续的过程中,似乎持续存在一系列启动因子,最终导致炎症反应终止的机制出现异常,在细胞增殖过程中,引发 DNA 损伤。
1.2 炎症反应
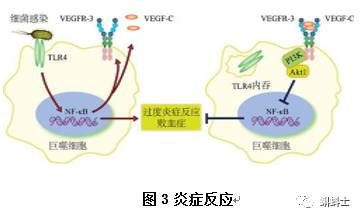
组织产生炎症反应有助于生理和病理过程,是一种正常的生理和病理过程,针对组织损伤,一种多因素的化学信号网络的响应,旨在“愈合”受影响的组织,这一过程涉及到受到激活的白细胞(中性粒细胞,单核细胞和嗜酸性粒细胞)的定向迁移,从静脉系统方面减少对组织的损害。中性粒细胞的作用机理被认为是协调这些受损伤组织的炎性细胞的聚集并产生细胞外基质(ECM),形成一个网络支架,导致成纤维细胞和血管内皮细胞发生增殖和迁移,从而提供了一种用于正常细胞反应的炎症微环境[7]。炎症因子 TNF-α(肿瘤坏死因子-α)控制炎症细胞的数量,以及介导炎症过程的其他的许多方面。TGF-β1 也是很重要的因子,从正面和负面来影响炎症和组织修复过程[8]。一个普遍的认识是,正常的炎症,例如,和伤口愈合相关的炎症通常是自限性的,但是中间任何的环节发生异常通常会导致细胞 DNA 的损伤和细胞增殖的紊乱,最终导致肿瘤的发生。
1.3 炎症细胞对肿瘤促进作用
炎症细胞对肿瘤的发展有着巨大的影响这一观点被很多人接受并认可。在肿瘤发生的早期,这些炎症细胞是强大的肿瘤启动因子,他们能够形成一个十分有吸引力的微环境,这个微环境进一步促进肿瘤的生长,促进基因组的不稳定以及促进血管的生成。炎症细胞,细胞因子以及炎症趋化因子,他们产生并影响整个肿瘤器官,影响肿瘤微环境中所有类型的细胞,调节它们的增殖,迁移和分化,包括肿瘤细胞、成纤维细胞和血管内皮细胞。在肿瘤的发生过程中,肿瘤细胞转向炎症机制,比如选择素与配体的相互作用,MMPs 的产生以及趋化因子,这些都有利于肿瘤的扩散和转移。这可能是肿瘤微环境拮抗并最终颠覆免疫细胞功能最重要的一步,免疫功能的紊乱和微环境的刺激有利于肿瘤的发生、发展。因此,病理情况下,炎症细胞的恢复和补充伴随着炎症因子的释放也可能会适得其反,肿瘤的发生可能是一种机体面对炎症刺激的一种反应。
2 慢性炎症对肿瘤的影响
2.1 慢性炎症可促进肿瘤的发生
慢性炎症可以通过病毒或细菌诱导,比如感染病原菌(例如幽门螺旋杆菌)能够导致胃炎,并增加罹患粘膜相关淋巴组织(MALT)淋巴瘤的风险[9,10]。乙型肝炎和丙型肝炎病毒(HBV,HCV),进而引起 HCC 的发生发展,幽门螺旋杆菌导致胃癌的发生等[9]。寄生虫(例如血吸虫)感染引起的慢性炎症可能导致膀胱癌,肝癌或结肠癌[11]。自身免疫性肝炎会增加肝癌发生的风险[12],以及炎性肠疾病(例如,溃疡性结肠炎,克罗恩病)会在某种程度上导致结肠癌的发生[13],PSC可导致胆管细胞癌(CCC)[14]。最近研究表明,饮食和遗传性肥胖也显示会促进肝炎和肿瘤的发生[15, 16]。
2.2 与慢性炎症相关的癌症
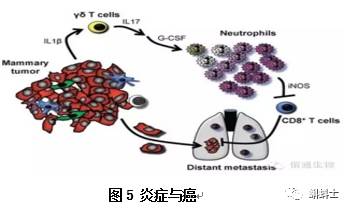
慢性炎症与恶性肿瘤的关联,最直接的一个例子是炎症性肠道疾病的患者发生结肠肿瘤,尤其是慢性溃疡性结肠炎和克罗恩病。在肝脏中,丙型肝炎感染诱发肝癌,膀胱癌的发生几率也会增加,结肠癌主要与与血吸虫病有关,而慢性幽门螺杆菌(革兰氏阴性菌的一种)已经被认定是胃癌发生的一个明确的致癌物,是引发胃癌的头号原因[17]。DNA 损伤导致慢性炎症被认为是肿瘤癌症发生的作用机制,炎症细胞诱导DNA 损伤能使巨噬细胞和 T 淋巴细胞表达巨噬细胞移动抑制因子(MIF)。MIF是一种多功能的细胞因子,通过克服 p53 的转录活性从而抑制 p53 的功能[18]。在浸润组织中,p53 调控功能的慢性循环可以增强细胞增殖和延长寿命,同时也创造了一个相对独立的微环境用于 DNA 损伤缺陷的响应,从而将潜在的致癌基因突变长时间积累,最终放大突变效应,引发肿瘤。
3肝的炎症与肝癌
3.1 肝脏的炎症
肝炎(即肝的炎症)指定的疾病范围很广。肝炎的临床范围很广,从没有症状或只有有限的症状,临床表现为为食欲不振,全身乏力和黄疸导致暴发性肝功能衰竭[19]。肝炎可以是自限性的或持续性的。急性和慢性肝炎主要从疾病症状表现的时间来定义,已是否具有较短时间或半年以上的持续时间区分。引起肝炎的病因很广,包括感染剂主要是嗜肝的其他病毒、细菌、原生动物等[20, 21],自身
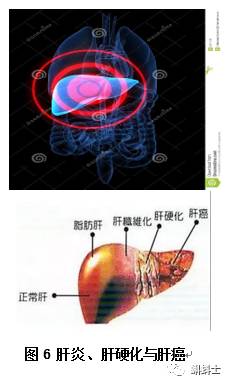
免疫,慢性胆汁淤积比如自身免疫性肝炎(AIH)[22],原发性硬化性胆管炎(PSC),代谢性疾病(如威尔森氏病),原发性胆汁性肝硬化(PBC)[23],非酒精性脂肪肝病(NAFLD),过多的营养毒性的损伤,包括酒精滥用[24],以及各种药物等[25-29]。肝炎的一个共同特征是炎症反应和肝组织的损伤共同存在。脱代偿性肝硬化的特征就是能够引发并发症,进而危及生命。肝硬化的另一个复杂因素是肝细胞癌(HCC)的发展。肝癌几乎可以从任何原因导致的肝硬化中转变而来,包括 AIH [26,27]和 PBC[27,30]。通过持续访问乙型肝炎携带者,肝硬化的转化似乎是肝细胞癌所必需的,包括 AIH 和 PBC。
3.2 肝细胞癌(HCC)
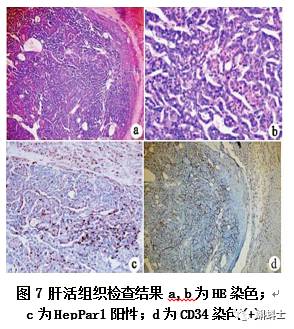
肝细胞癌(HCC)是人类最常见的癌症之一,每年都会造成全球数百万人丧生。尽管随着科学的进步,肝癌的诊断和治疗技术取得很大的进步,但由于具有转移复发性,肝癌依旧是一个高度致死性肿瘤。肝细胞癌是炎症相关肿瘤的一个确切的例子,慢性炎症状态似乎是肝癌发生和发展所必需的[31]。肝脏的炎症反应具有自限性,并且其持续性取决于病因,比如肝脏的健康状态,毒素或病原体的浓度,以及暴露于毒素或感染的时间。如果免疫系统杀灭病原体或暴露的毒素是短暂的,急性肝炎便会得到消解并且受影响的肝组织会自我恢复。然而,在许多情况下,肝损伤是慢性的,慢性肝损伤会导致慢性肝炎和肝细胞的死亡,以及代偿性增生,这些都会影响肝脏的再生,并且有可能进一步促进肝损伤、肝纤维化、肝硬化和肝癌的发生。
肝癌已经成为世界上最常见的癌症之一,在一些非洲和亚洲国家,HCC 已成为癌症死亡的最普遍的原因[32,33]。在以往的科学研究中,临床上药物治疗肝癌患者的成功性是有限的,目前,肝移植是提高肝癌患者生存时间最有效的方法[34]。科学研究者也一直在努力研究新药物,近年来,各种抑制剂分子已进入临床实践阶段。比如,索拉非尼是一种新的治疗药物,能够抑制前血管的生成,以及肿瘤性受体酪氨酸激酶的活性(如 c-kit;VEGFR-1、VEGFR-2、VEGFR- 3;PDGFRβ),它对于肝癌治疗的疗效已经在临床试验中得到证明[35, 36]。此外,其它治疗剂,如 Regorafenib(BAY73-4506),科学研究者正在评估其作为抗肿瘤疗法的潜力。
参考文献
[1] Balkwill F, Mantovani A.Inflammation and cancer: back to Virchow?[J]. Lancet, 2001, 357(9255): 539-545.
[2] Dvorak H F. Tumors: wounds that donot heal. Similarities between tumor stroma generation and wound healing[J]. NEngl J Med, 1986, 315(26): 1650-1659.
[3] Arend P. Ancestral gene and"complementary" antibody dominate early ontogeny[J]. Immunobiology, 2013, 218(5):755-761.
[4] Pardoll D M. Spinning molecularimmunology into successful immunotherapy[J]. Nat Rev Immunol, 2002, 2(4): 227-238.
[5] Rous P, Kidd J G. ConditionalNeoplasms and Subthreshold Neoplastic States : A Study of the Tar Tumors ofRabbits[J]. J Exp Med, 1941, 73(3): 365-390.
[6] Mackenzie I, Rous P. TheExperimental Disclosure of Latent Neoplastic Changes in Tarred Skin[J]. J ExpMed, 1941, 73(3): 391-416.
[7] Schleimer R P. Inflammation: Basicprinciples and clinical correlates edited by John Gallin, Ira Goldstein andRalph Snyderman, Raven Press, 1987. $219.00 (xvii + 995 pages) ISBN 088167 3447[J]. Immunol Today, 1988, 9(10): 327.
[8] Moustakas A, Pardali K, Gaal A, etal. Mechanisms of TGF-beta signaling in regulation of cell growth anddifferentiation[J]. Immunol Lett, 2002, 82(1-2): 85-91.
[9] Llovet J M, Beaugrand M.Hepatocellular carcinoma: present status and future prospects[J]. J Hepatol,2003, 38 Suppl 1(S136-149.
[10] Kearney D J, Liu C F, Crump C, etal. The effect of a Helicobacter pylori treatment strategy on health care expenditures in patients with peptic ulcer disease and dyspepsia[J]. Am J Gastroenterol, 2003, 98(9): 1952-1962.
[11] Mostafa M H, Sheweita S A,O'Connor P J. Relationship between schistosomiasis and bladder cancer[J]. Clin Microbiol Rev, 1999, 12(1):97-111.
[12] Zhou D S, Xu L, Luo Y L, et al. Inflammation scores predictsurvival for hepatitis B virus-relatedhepatocellular carcinoma patients after transarterial chemoembolization[J]. World J Gastroenterol,2015, 21(18): 5582-5590.
[13] Pohl C, Hombach A, Kruis W.Chronic inflammatory bowel disease and cancer[J]. Hepatogastroenterology, 2000, 47(31): 57-70.
[14] Rossi R E, Conte D, Massironi S.Primary sclerosing cholangitis associated with inflammatory bowel disease: an update[J]. Eur J Gastroenterol Hepatol, 2016, 28(2): 123-131.
[15] Fan J G, Farrell G C. Epidemiologyof non-alcoholic fatty liver disease in China[J]. J Hepatol, 2009, 50(1): 204-210.
[16] Park E J, Lee J H, Yu G Y, et al.Dietary and genetic obesity promote liver inflammation and tumorigenesis byenhancing IL-6 and TNF expression[J]. Cell, 2010, 140(2): 197-208.
[17]Ernst P B, Gold B D. The diseasespectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcerand gastric cancer[J]. Annu Rev Microbiol, 2000, 54(615-640.
[18]Hudson J D, Shoaibi M A, Maestro R,et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity[J]. JExp Med, 1999, 190(10): 1375-1382.
[19] Centeno B A. Pathology of livermetastases[J]. Cancer Control, 2006, 13(1): 13-26.
[20] Dienstag J L. Hepatitis B virusinfection[J]. N Engl J Med, 2008, 359(14): 1486-1500.
[21] Wedemeyer H, Manns M P.Epidemiology, pathogenesis and management of hepatitis D:update and challenges ahead[J]. Nat Rev GastroenterolHepatol, 2010, 7(1):31-40.
[22] Krawitt E L. Autoimmunehepatitis[J]. N Engl J Med, 2006, 354(1): 54-66.
[23] Kaplan M M, Gershwin M E. Primarybiliary cirrhosis[J]. N Engl J Med, 2005, 353(12): 1261-1273.
[24] Lucey M R, Mathurin P, Morgan T R.Alcoholic hepatitis[J]. N Engl J Med, 2009, 360(26): 2758-2769.
[25] Tujios S, Fontana R J. Mechanismsof drug-induced liver injury: from bedside to bench[J]. Nat Rev GastroenterolHepatol, 2011, 8(4): 202-211.
[26] Matsumoto K, Onoyama T, Kawata S,et al. Hepatitis B and C virus infection is a risk factor for the developmentof cholangiocarcinoma[J]. Intern Med, 2014, 53(7): 651-654.
[27] Friedmann S, Dantes A, Amsterdam A. Ovariantranscriptomes as a tool for a global approachof genes modulated by gonadotropic hormones in human ovarian granulosacells[J]. Endocrine, 2005, 26(3): 259-265.
[28] Stojsavljevic S, Gomercic PalcicM, Virovic Jukic L, et al. Adipokines and proinflammatory cytokines, the keymediators in the pathogenesis of nonalcoholic fatty liver disease[J]. World JGastroenterol, 2014, 20(48): 18070-18091.
[29] Yeoman A D, Al-Chalabi T, Karani JB, et al. Evaluation of risk factors in the development of hepatocellularcarcinoma in autoimmune hepatitis: Implications for follow-up and screening[J].Hepatology, 2008, 48(3): 863-870.
[30] Cavazza A, Caballeria L, Floreani A, et al. Incidence, risk factors,and survival of hepatocellular carcinomain primary biliary cirrhosis: comparative analysis from two centers[J]. Hepatology, 2009, 50(4): 1162-1168.
[31] Shinkawa H, Nakai T, Tamori A, etal. Hepatocellular carcinoma (HCC) recurring 10 years after clearance of hepatitis B surface antigen and 20 years after resection of hepatitis B virus-related HCC[J]. Int JClin Oncol, 2008, 13(6): 562-566.
[32] El-Serag H B, Rudolph K L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis[J].Gastroenterology, 2007, 132(7): 2557-2576.
[33] Sherman M. Epidemiology ofhepatocellular carcinoma[J]. Oncology, 2010, 78 Suppl 1(7-10).
[34] Dutkowski P, De Rougemont O,Mullhaupt B, et al. Current and future trends in liver transplantation in Europe[J].Gastroenterology, 2010, 138(3): 802-809 e801-804.
[35] Llovet J M, Ricci S, Mazzaferro V,et al. Sorafenib in advanced hepatocellular carcinoma[J]. N Engl J Med, 2008,359(4): 378-390.
[36] Jaal J, Kase M, Minajeva A, et al.VEGFR-2 Expression in Glioblastoma Multiforme Depends on Inflammatory Tumor Microenvironment[J]. Int JInflam, 2015, 2015(385030.










