一开始拿到三代测序的下机数据时,一脸懵逼。怎么这么多文件?怎么没有raw data, clean data了?这些乱七八糟的文件都是干嘛的?今天我们就来聊聊三代测序的下机数据都有哪些,以及他们具体的格式是怎么样的(以sequel 平台为主)。
测序过程
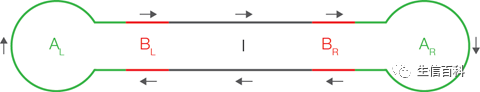
SMRTbell
A adapter通用接头,两端的接头可以一样也可以不一样
B barcode(客户自己设计)
I insert 插入片段,即我们测序的目的片段
由于SMRTbell是环状的,测序过程是边合成边测序,因此可以沿着新链合成的方向不停地读取序列,读取一圈又一圈,直到聚合酶累趴下了…
测序结果
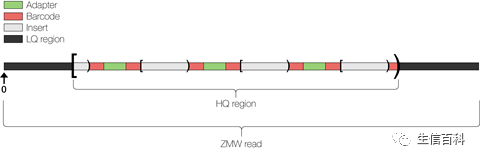
根据SMRTbell的形状以及测序的过程,我们容易知道,测序出来的reads如上图所示,由接头序列, 条码序列, 插入序列间隔线性分布,即ABIB-ABIB—ABIB-ABIB—…(A: adapter, B: barcode, I: insert)
ZMW read 是测序出来的完整结果,也即是polymerase read,聚合酶合成过的所有的序列。
PostPrimary 分析后输出HQ region,由ZMW read 去除两端低质量区域得到。
收到的测序文件
RS II
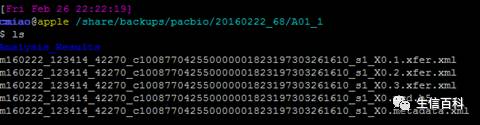
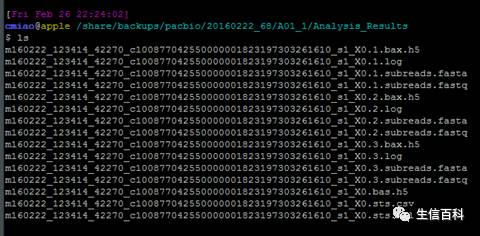
RS II 的 文件格式如上图所示,由于本篇文章主要描述sequel平台的下机数据, RS II的文件格式就不详聊了,放一个相关的链接,大家有兴趣可以参考。
参考网址:
https://www.cnblogs.com/freemao/p/5222151.html
Sequel
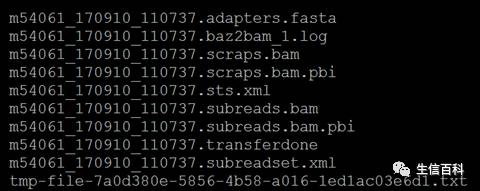
在下机文件中,主要有三类文件,bam 文件,bam.pbi 文件,以及xml文件。
当我们习惯性的去寻找熟悉的fastq格式文件做分析时,忽然发现找不到了,因为在sequel平台中bam 文件成为了它的替代者,因为其更节约储存空间。这是文件格式的一个重大更新。
用于后续分析的文件一般是.subreads.bam,这等同于RS II 中的.subreads.fastq
下面仔细聊聊三类主要文件的具体格式,以及他们分别干什么活的。
Pacbio 的BAM 文件格式
我们平常见到的bam文件大多是比对结果文件,例如用重测序分析中BWA生成的bam文件就是reads与基因组的比对文件。但pacbio的下机文件是没有与基因组进行过比对过的,其主要作用就是储存序列。
Bam文件主要分为两个部分,头一部分是Header,储存测序的相关信息,另一部分也即是文件的主要部分是records,这里头保存了我们的序列信息。我们这里就以subreads.bam文件为例,分析下bam文件的具体格式。
可以用samtools view 命令查看bam文件
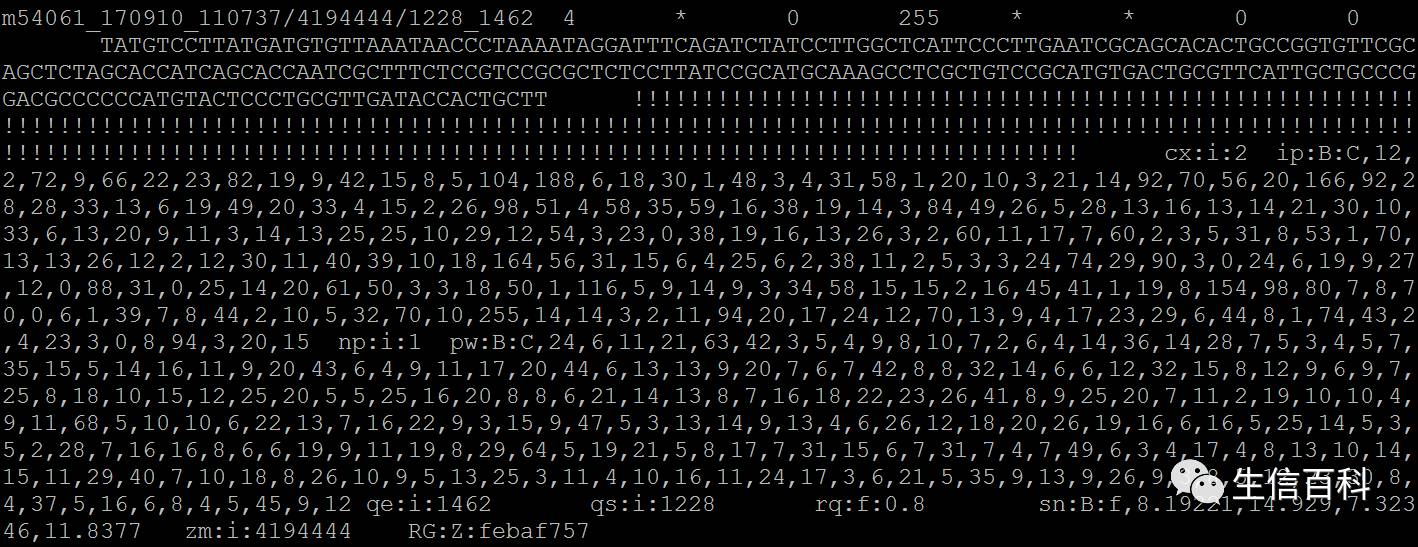
第一列:reads信息
{movieName}/{holeNumber}/{qStart}_{qEnd}
[对于CCS:{movieName}/{holeNumber}/ccs]
MovieName 是cell的名字,holeNumer是ZMW孔的编号,qStart和qEnd是subreads相对于ZMW reads的位置。
第二列 (sum of flags):比对信息 均为4 代表没有比对上,也表明了bam文件只储存了序列信息,而没有比对信息。
第三列 (RNAM):参考序列 值为 ,代表无参考序列
第四列 (position) : 比对上的第一个碱基位置 0
第五列 (Mapping quality) : 比对质量分数 255
第六列 (CIGAR值) : 比对的具体情况
第七列 (MRNM, ) : mate 对应的染色体
第八列 (mate position) : mate对应的位置 0
第九列 (ISIZE, Inferred fragment size) : 推断的插入片段大小 0
第十列 (Sequence) : 序列信息 具体的ATCG
第十一列 (ASCII码) : 碱基质量分数 ASCII+33
第十二列 : 可选区域 记录Reads 的总体属性包括信号长度,信号强度等信息。
BAM 文件分别都是些什么?

zmws.bam 以及ccs.bam似乎公司并不一定会提供