前言
美国药典(USP)将增加一个新的通则<1236>Solubility Measurements。这一新的章节概述了与溶解度测定相关的概念和方程,还简要介绍了用于评估药物溶解度的典型实验方法,并讨论了溶解度的测定方法,以获得生物相关的用于人和兽药产品的特定物种的溶解度。
USP<1236>溶解度的测定
一种物质在另一种物质中的溶解度是热力学平衡中两种纯物质之间分子混合程度的量度。饱和溶液的组成,用在指定溶剂中的指定溶质的比例来表示,代表了溶解度的热力学极限。溶解度可以用质量摩尔浓度、摩尔分数、摩尔比、重量/体积比、重量/重量比等浓度单位表示。准确测定药物的水溶性对于理解药物制剂的质量控制和药物传递问题是非常重要的,受药物物理化学性质(例如表面积、颗粒大小、晶型)、介质的性质(例如pH、极性、表面张力、添加表面活性剂、助溶剂、盐)、以及溶解度测定参数的控制(例如温度、时间、搅拌法)等因素的影响。在溶解度测定中控制这些因素是获得准确、可靠的物质平衡溶解度的关键。
本章将首先讨论与溶解度测定有关的概念和方程。理解这些关系是准确评估溶解度的基础。接下来将简要介绍用于评估药物溶解度的典型实验方法。最后,讨论了溶解度测量以获得生物相关的溶解度(人用产品)和物种依赖性溶解度(兽用产品)。
背景
热力学平衡与溶解度
混合吉布斯自由能,ΔGmix,决定了两个化合物混合形成溶液的可能性和程度。

其中,ΔHmix是混合焓,表明混合是吸热还是放热过程,T是热力学温度,开尔文;ΔSmix是混合熵,是纯系统的混乱程度的度量。如果吉布斯自由能的变化是负的,混合将是自发的,当达到平衡时,ΔG等于零。
混合焓ΔHmix,是溶质的焓和溶剂的焓之和与混合物焓间的差值,即:
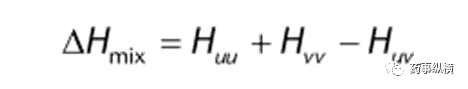
其中u是溶质,v是溶剂。如果焓变是负的,混合将放热,反映出附着力作用比内聚力作用更强,有利于混合。一个理想的系统,ΔHmix是零,因为理想的溶质和溶剂之间的相互作用是相同的。因此,对于一个理想的系统:

其中Xu是以摩尔分数表示的溶质浓度,R是气体常数。当一个结晶固体与饱和溶液处于平衡状态时,固体和饱和溶液的吉布斯自由能是相等的。同样,溶液的吉布斯自由能可以认为是理想溶剂和理想溶质液体混合的结果,因此:
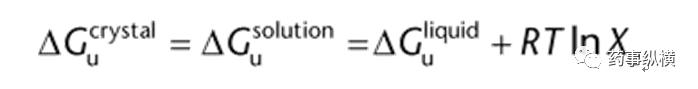
根据Kirchhoff’s定律,不可逆过程的能量等于一系列可逆过程的能量。因此,固体的溶解过程相当于将固体加热到熔点,然后将液体溶质与溶剂混合,最后将溶液冷却下来。利用Kirchhoff’s定律和上述方程,可以描述为(1):

Xu是理想溶质在25℃下的摩尔分数,MP是结晶固体的熔点(℃)。这个方程式说明了结晶固体的熔点与摩尔溶解度的关系。熔点较高的晶体化合物具有较高的内聚力,必须克服,才能将其转化为液体,才可与溶剂混合,作为理想的溶质。
水溶解中溶解度的估算方法
Yalkowsky证明了(1、2)一个相对简单的一般溶解度方程(GSE)可以用于估算化合物在水中的固有溶解度。

其中S0是内在的溶解度(未解离分子),MP是结晶固体的熔点(℃),KOW是正辛醇-水分配系数,水温度25℃。一般溶解度方程表明,具有较高熔点的化合物以及具有较高亲油性的化合物的水溶性将降低。一般溶解度方程中正辛醇-水分配系数的对数说明了理想溶液和水溶液混合焓的不同(2)。如果pKa是已知的,一般溶解度方程也可以与Henderson-Hasselbalch方程相结合用来预测离子化合物的溶解度(见影响溶解度和溶解度测定的影响因素,pH值)。
尽管一般溶解度方程很简单,但使用它时需要测量熔点和分配系数(或离子化合物的pKa)。有几种计算机软件用于基于结构的化合物(3)的分配系数和pKa值的估算,但熔点不能。开发来预测水溶液的计算方法依赖于分子的训练集,以寻找与结构(例如,分子量、溶剂可及表面积、可旋转的键的数量等)(3)。这些计算方法的成功往往局限于类似训练集的分子。这些计算方法足以在预筛选合成候选者中提供帮助,但不足以取代实验溶解度测定。
影响溶解度和溶解度测定的因素
pH值的影响
离子化的酸和碱的溶解度是pH依赖性的,因为带电部分具有较高的水溶性。Henderson–Hasselbalch方程将溶解度的增加与溶液pH值相对于电离酸或碱的pKa或pKb联系起来。
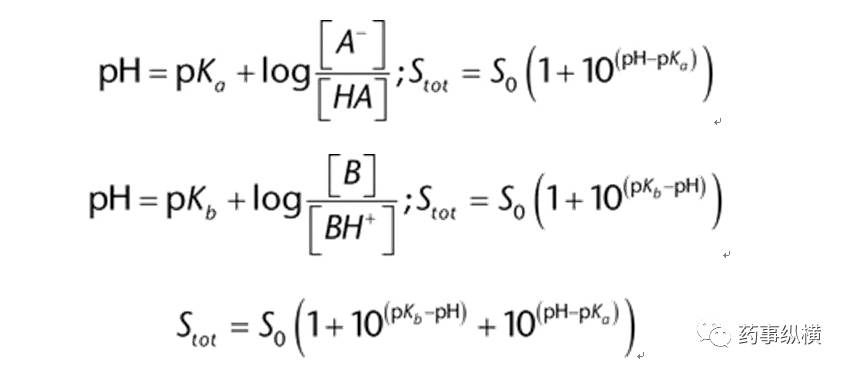
[HA]是未离解弱酸的摩尔浓度,[A-]是酸的的共轭碱的摩尔浓度,pKa=-log(Ka),Ka是酸离解常数。
分子在pH值低于5.6和高于11.7时电离,在这两个pH值之间时呈中性。分子没有离子化时,溶解度等于固有溶解度。对于离子化分子,随着pH值的变化,溶解度呈对数增加。成盐后将限制在较低或较高pH值下的溶解度(见图1)。如果用来调节pH值的酸有助于增加盐的反离子,那么同离子效应会随着这个反离子浓度的增加而进一步抑制盐的溶解度(见图1)。如果盐在较高pH值下溶解,最初盐可能会过饱和,但最终将会在这个pH下因为溶解度小而形成沉淀(4)。

图1:pH对离子化合物溶解度的影响。当分子没有离子化时,溶解度相当于固有溶解度。对于电离分子,随着pH值的变化,溶解度呈对数增加。盐的溶解度限制了低pH值下的溶解度,如果用于调节pH值的酸对盐的反离子有一定贡献,那么,同离子效应会随着这个反离子浓度的增加而抑制盐的溶解度(图中pH值<2)。
盐和反离子的影响
可电离的化合物也可以用相反的带电离子(4)来形成盐。在溶液中,在带电的反离子存在的情况下,带电分子的溶解度由溶解度积给出,描述这个平衡反应的方程如下:

盐在溶液中有限的溶解度如图1所示。由于盐的形成,带电分子的实际溶解度被认为是稳定的(在低pH值时),而不是像Henderson-Hasselbalch方程所预测的那样继续增加。因为溶度积,KSP,是一个常数,离子化基团的溶解度可能会随着调节pH值的带相反电荷的反离子浓度增加而下降。随着离子对浓度的增加带电分子的溶解度降低的现象称为同离子效应(4)。经常看到,当氯化氢(HCl)用来降低pH值时,氯盐的溶解度会由于氯浓度的增加而降低(如在pH<2)。虽然在图1中没有说明,盐的溶解度也可能在曲线的碱一侧(例如酸部分的钠盐)受到限制,高pH值时,同离子效应同样会影响到盐的溶解度(例如氢氧化钠)。
潜溶剂的影响
对于许多药物,水通常是一个溶解性差的溶剂。但水与其他溶剂可混溶,这些溶剂可为这些物质提供良好的溶解性(如乙醇、丙二醇、聚乙二醇等)。根据对数-线性模型(1),在两个混合溶剂中,溶质的对数可以通过线性方程进行表示。这种关系如图2所示。当溶解度曲线被转换成线性时,很明显,即使是混合溶剂中的低浓度的溶剂(通常是水),也可以大大降低溶质的溶解度。因此,由于溶解度发生显著变化,含有潜溶剂的溶液在稀释时药物特别容易发生沉淀。
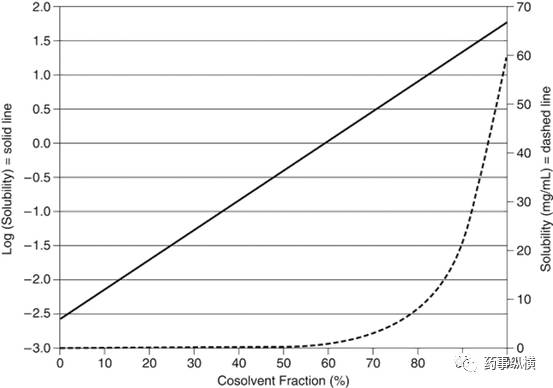
图2:Yalkowsky等人的对数-线性模型的说明(1)。溶解度的对数与潜溶剂体积分数呈线性关系。当用线性绘制时,很明显,溶解度随着加入到溶解性好的溶剂中的溶解性差的溶剂的增加呈指数形式下降。
表面活性剂的影响
在临界胶束浓度(CMC)之上,溶液中胶束的数量会随着表面活性剂浓度的增加线性增加。如果一种药物可以嵌入在胶束中,其溶解度将随着胶束数量的增加而线性增加(见图3)。
如图3所示,表面活性剂存在下的溶解度是水相中溶解的量加上胶束溶解的量。胶束比溶质更大,扩散速度比溶质更慢。在胶束的存在下的药物的传递是由于在溶液中吸收了游离的药物,以及由微粒子介导的转运(5,6)。因此,通过表面活性剂的溶解可能不会导致与水溶性的增加直接成正比的药物传递的增强(5、6)。

图3:通过表面活性剂使溶解度增加。溶解需要胶束的形成。在CMC以下增加表面活性剂,会使表面活性剂在溶剂中溶解,没有发生溶解度增加。在CMC之上,溶解度呈线性增加。这种线性增加的斜率表明胶束的溶解效率。
络合剂的影响
络合剂可与低溶解度物质形成络合物,从而提高其溶解度。不管配体和溶质在这些络合物中的比例如何(例如,1:1、2:1、3:1等等),随着络合剂浓度的增加,溶解度将呈线性增加(7)。这与表面活性剂的溶解度非常相似,但不存在所需的络合剂的最小浓度。具有高稳定性常数的络合物可以紧密结合溶质,从而提高溶液稳定性。环糊精常被用来与药物形成络合物来提高溶解度,但是环糊精是非常亲水的,具有很高的分子量(大于970g/mol),并且不容易渗透到组织中(7)。因此,强结合的环糊精络合物可能会延缓药物的传递,因为只有未结合的药物才能穿透组织(7)。
表面积的影响(溶解速率)
Noyes–Whitney方程为:

D是溶质的扩散系数,A是溶质粒子的表面积,h是扩散层的厚度,Cs是溶质的饱和溶解度,C是t时间溶质的浓度。对于小颗粒,扩散层厚度h,通常认为等于粒子半径(=½d)。对于球形颗粒,表面积A,可以表示为一个总质量M的函数,密度ρ,粒径d。
物质的溶解速率不会影响平衡溶解度,但会影响达到平衡的速度。这个方程表明,较小的粒子会有更大的表面积,并且会更快地溶解。为了尽可能快地达到平衡溶解度,应尽可能地保持大的表面积(例如更小的粒子)和尽可能小的扩散层厚度(例如良好的混合)(8)。
表面能的影响(纳米粒子)
当粒子尺寸接近纳米粒子的范围时,粒子的表面能量可能影响溶解度。根据Kelvin方程,由于表面能量对系统总吉布斯自由能的影响,较小的粒子比较大的粒子具有更高的溶解度。Kelvin方程将其量化为:
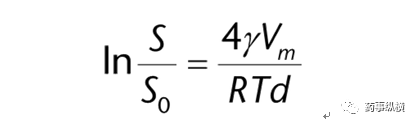
其中R是气体常数,T是温度,𝛾是溶质的表面能(在溶质-溶剂界面),Vm是溶剂的分子体积,S0是一个无限大颗粒的溶解度,S是直径为d的粒子的表观溶解度。通常,这种对溶解度的影响只会对小于1微米的粒子产生影响。
小颗粒和大颗粒之间的溶解度差会导致所谓的Ostwald熟化。小颗粒溶解,导致在溶液中相对于大颗粒的溶解度过饱和,这导致在较大颗粒表面重结晶。较大的颗粒长大,而较小的颗粒溶解,导致悬浮颗粒的平均粒径增加(9)。
实验方法
平衡溶解度测定方法
饱和摇瓶法
摇瓶法是基于40年前开发的相溶解度技术,至今仍被大多数人认为是最可靠和最广泛使用的溶解度测定方法(10 - 15)。当需要测定平衡溶解度时,应使用摇瓶法。其他方法可以用来评价表观溶解度,但不适合评价真正的平衡溶解度。
选择溶解度测量的溶解介质应与其应用相关,并控制表面活性剂的类型和浓度、缓冲液的离子强度、以及在缓冲液中存在的反离子的类型。当结果旨在预测吸收或生物利用度时,建议使用一种生物相关的介质溶液(参见生物相关介质中的溶解度测定)。当结果旨在支持溶出方法开发时,建议使用溶出介质。出于研究目的,当该化合物的pH依赖性正在评估时,建议缓冲液在一个宽的pH范围内控制离子强度和反离子类型(例如,Britton–Robinson or Sörensen缓冲液)。
样品制备:待测物质通常是将过量固体加入到溶解介质中来制备的,这种溶解介质装在一个具塞瓶中。瓶中的介质的量不需要精确测量。建议将固体加到溶解介质中,大约1-2mg/ml,要超过估计的溶解度(对于低溶解度化合物,1-2mg/mL的浓度可能已足够)。固体的表面积可以在加入前通过研磨来增加(例如,用研钵和杵)或在加入到介质中后通过对样品进行超声来增加。
溶液的平衡:为了便于固体溶解,混悬液应混合或搅拌24h。混悬液的温度在整个溶解阶段应该很好的进行控制 (±0.5℃)。在溶解阶段后,建议将多余的固体完全沉淀。取上清液时应避免混入任何不溶解的固体,因为这会显著地影响溶解度的结果。如果沉淀不完全,不能提供一个澄清的上清液进行分析,那么可以使用离心法。混悬液的温度在沉降和离心步骤中也必须很好的控制(±0.5℃),应与溶解时的温度相同。
当对多个样品经过不同的平衡时间进行测定而产生相等的结果时,说明达到了饱和(平衡)。为了确认表观溶解度是平衡溶解度,建议同样的混悬液采用同样方式重新平衡(例如,再增加24h)。
溶液的分析:上清液在分析前可能需要稀释,以保证在分析方法的线性范围内,并且避免产生可能的沉淀。可以采用UV法或HPLC法测定溶液浓度。HPLC法的优点是可以测定不稳定药物的有关物质(12、16)。
溶解度结果的报告:在抽取样品进行分析时,应记录上清液的pH值(在溶解度测量的温度下)。应报告在平衡步骤结束时的pH值和温度下的溶解度值(11)。建议在溶解度测量结束时分析混悬液中过剩的固体,以验证固体形态没有改变。PXRD法或DSC法可以用来评价固体。所报告的平衡溶解度的精密度应该反映在随后的时间点上测量之间的一致程度,而不是溶解度测定的精密度。将平衡溶解度报告为最后时间点的溶解度±最后两个溶解度测量值的差值。
表观溶解度测定方法
电位滴定法
溶解度测定中的酸碱电位滴定法是基于由沉淀引起的滴定曲线中间的一个特征位移。滴定时,将准确体积的标准酸或碱加入到一个含有可电离物质和0.15M氯化钾(为了增加测量的准确性)的溶液中。通过氩气Sparging(一种技术,将化学惰性气体如氮气、氩气或氦气通入到液体中)可以防止大气中的二氧化碳对pH值产生影响。使用一个玻璃电极对pH值进行连续监测。通过酸/碱消耗的体积和pH值得到滴定曲线(12)。
比浊法
比浊法是将化合物溶解在有机溶剂中,通常是二甲基亚砜(DMSO)。每隔1分钟时间,将上述溶液加入到pH7的缓冲液中。通过光散射法对浊度进行第一次检测后,再加入上述溶液。随后,可将加入的体积与浊度作图,然后向后外推至沉淀开始的点作为估计的溶解度。这种方法可以用来测量多达50~300个样品/日。这种方法的缺点包括:DMSO的使用可提高溶解度(作为助溶剂)至一个未知的程度,实验的持续时间短,这导致了一个动力学而不是热力学溶解度估计,析出固体的晶体形态是不知道的,除非它是从悬浮液中分离和溶液中进行表征的。DMSO会稳定形成过饱和溶液,动态测量方法容易预测药物的表观溶解度(12)。
溶解度测定的小型化、高通量和自动化
溶解度不仅在处方前研究中起重要作用,而且在药物开发过程中先导化合物的选择和优化中也起着重要作用。因此,希望有能够以尽可能少的化合物来确定表观溶解度的方法,以及具有必要的高通量特性,使得该方法可用于支持化合物的优化(16)。为此,Glomme等人(12)开发了一种小型化的摇瓶法。该方法基本相当于传统的摇瓶法,但Glomme的小型化法允许使用非常少的溶剂(例如0.5-2ml,这取决于所使用的设备)。
在生物相关介质中的溶解度的测定
除非另有说明,在生物相关溶解度测量中,溶解介质的温度应控制在37±0.5℃。生物相关溶解度测量应遵循摇瓶法,包括在24h的溶解度测量和至少一个额外的时间点,以确认达到平衡的精度。加入到溶解度介质中的药物的盐形式会影响达到平衡的方法。因此,盐和相同药物游离碱的溶解度不应该被认为是一致的,除非开始就通过不同的结晶固体进行独立的测定来证明。
人空腹模拟胃液(FASSGF)(17)
pH:1.6(37℃)
介质组成:

人进食模拟胃液 (FESSGF) (17)
pH:5(37℃)
介质组成:
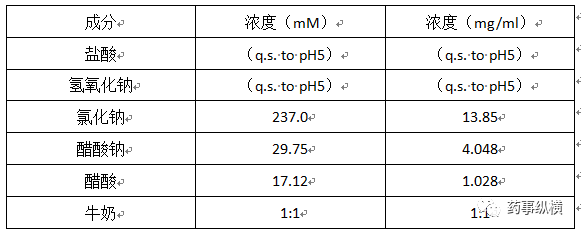
制备缓冲液,与牛奶混合1:1。如果必要的话,调整pH值为5。
人空腹模拟肠液 (FASSIF-V2) (17)
pH:6.5(37℃)
介质组成:

人进食模拟肠液 (FESSIF-V2) (17)
pH:5.8(37℃)
介质组成:

人模拟结肠液(SCOF2) (18)
pH:5.8(37℃)
介质组成:

其他动物物种(如狗和牛)的生物相关介质略,详见USP<1236>原文。
术语表
备注:在本章的上下文中,提供了下列定义,以阐明这些术语的使用。这些定义并不打算取代或否定USP和NF中其他地方的定义。
附着力:是不同分子(溶质-溶剂)之间的热力学相互作用。
内聚力:是同一分子(溶质-溶质,溶剂-溶剂)的热力学相互作用。
表观溶解度:是溶质在溶剂体系中的经验确定溶解度。由于短暂的过饱和或不完全溶解或达到平衡的时间不足,表观溶解度可能高于或低于平衡溶解度。
固有溶解度:是不带电的溶解度(中性)。固有溶解度只能在pH值范围内精确测量,在这个范围内,物种的分布由不带电荷的分子支配。对于某些化合物,可能无法直接测量固有溶解度,必须通过溶解度数据的拟合来确定。
溶解:是在热力学平衡(例如,溶质和溶剂形成均匀混合溶液)中接近溶解度极限的非平衡过程。溶解速率会影响达到平衡所需的时间,但不会影响最终平衡溶解度。
溶解度:是在热力学平衡条件下的浓度极限,溶质可以均匀地混合到溶剂中。这可以称为平衡(饱和)溶解度,以区别于表观溶解度。溶解度可以用质量摩尔浓度、摩尔分数、摩尔比、重量/体积比、重量/重量比等浓度单位表示。
参考文献
[1]Yalkowsky SH. Solubility andSolubilization in Aqueous Media. ACS and Oxford University Press; 1999:464.
[2]Ran Y, Yalkowsky SH.Prediction of drug solubility by the general solubility equation (GSE). J ChemInf Comput Sci. 2001;41(2):354–357.
[3]Jorgensen WL, Duffy EM.Prediction of drug solubility from structure. Adv Drug Deliv Rev.2002;54:355–366.
[4]Serajuddin AT. Saltformation to improve drug solubility. Adv Drug Deliv Rev. 2007;59(7):603–616.
[5]Johnson KA, Westermann-ClarkGB, Shah DO. Controlled release of steroids through microporous membranes withsodium dodecyl sulfate micelles. Pharm Res. 1989;6(3):239–243.
[6]Poelma FGJ, Breas R, TukkerJJ. Intestinal absorption of drugs. III. The influence of taurocholate on thedisappearance kinetics of hydrophilic and lipophilic drugs from the smallintestine of the rat. Pharm Res. 1990;7(4):392–397.
[7]Loftsson T, Jarho P, MassonM, Jarvinen T. Cyclodextrins in drug delivery. Expert Opin Drug Deliv.2005;2:335–351.
[8]Sheng JJ, Sirois PJ,Dressman JB, Amidon GL. Particle diffusional layer thickness in a USPdissolution apparatus II: a combined function of particle size and paddlespeed. J Pharm Sci. 2008;97(11):4815–4829.
[9]Verma S, Kumar S, Gokhale R,Burgess DJ. Physical stability of nanosuspensions: investigation of the role ofstabilizers on Ostwald ripening. Int J Pharm. 2011;406:146–152.
[10]Baka E, Comer JEA, Takács-Novák K. Study of equilibrium solubilitymeasurement by saturation shake-flask method using hydrochlorothiazide as modelcompound. J Pharm Biomed Anal. 2008;46:335–341.
[11]Völgyi G, Baka E, Box KJ, Comer JEA, Takács-Novák K. Study ofpH-dependent solubility of organic bases. Revisit of Henderson–Hasselbalchrelationship. Anal Chim Acta. 2010;673:40–46.
[12]Glomme A, März J, Dressman JB. Comparison of a miniaturized shake-flasksolubility method with automated potentiometric acid/base titrations andcalculated solubilities. J Pharm Sci. 2005;94(1):1–16.
[13]Box KJ, Völgyi G, Baka E, Stuart M, Takács-Novák K, Comer JEA.Equilibrium versus kinetic measurements of aqueous solubility, and the abilityof compounds to supersaturate in solution—a validation study. J Pharm Sci.2006;95(6):1298–1307.
[14]Brittain HG. Solubility methods for the characterization of new crystalforms. In: Adeyeye MC, Brittain HG, eds. Preformulation in Solid Dosage FormDevelopment. London:Infroma; 2008:323–346.
[15]Higuchi T, Connors KA. Phase solubility techniques. Adv in Anal Chem andInst. 1965;4:117–212.
[16]Tong W-Q (Tony). Practical aspects of solubility determination inpharmaceutical preformulation. In: Augustijns P, Brewster ME, eds. SolventSystems and Their Selection in Pharmaceutics and Biopharmaceutics. NewYork:Springer; 2007:137–149.
[17]Jantratid E, Dressman J. Biorelevant dissolution media simulating theproximal human gastrointestinal tract: an update. Dissolution Tech. 2009:21–25.
[18]Marques MRC, Loebenberg R, Almukainzi M. Simulated biological fluidswith possible application in dissolution testing. Dissolution Tech. 2011:15–28.
[19]M Arndt, et al. Dissolution media simulating the proximal caninegastrointestinal tract in the fasted state. Eur J Pharm and Biopharm.2013;84:633–641.
声明:药事纵横小编因个人学习与研究需要翻译本文,请尊重药事纵横小编劳动成果,转载本文请先获得药事纵横授权,并注明来源、作者和译者,否则一律视为恶意侵权,侵权必究!
更多干货尽在药事纵横网站,地址:www.pharmaguider.cn,可点击原文链接一键直达。

药事纵横是一个开放,由自愿者组成的团体,现有成员13名,分别为 Voyager88(魏利军),雷诺岛,三分话,Herman,Mzwinsunny,文竹,duke,巉巉之石,占小兵,ISAL,海角边,yhqqqqq,鲁礼炎,欢迎有志之士加入我们团队。投稿、加专业微信群【合成、制剂、分析、注册、BD、一致性评价】请加微信442015666,QQ群:22711679










