
我为什么要做生信分析——这个问题回答起来其实很简单,因为如果你做了一个芯片测序,你得到的是下面这些东西:

相信这应该不是你想要的结果吧?
你想知道的是应该是
究竟是哪一个或者哪一群基因在某个生物学过程中起到了重要的作用
,而要得到这样一个可信的结论,是需要进行重重的生物信息学分析的。
生物信息学分析大致可以分为
三个境界
:
1、
只会机械的套用已有的方法
,对算法和原理一无所知,无法运用结果解释分析生物学问题;
2、
了解生信检验的基本原理
(作者在发明它时,最初的构想、原型、启发),可以根据实际情况选择不同的分析算法,采用最优解,能够解释生物学问题;
3、
能够自由的组合、拼接已有的算法,必要时创造想要的算法
。
回到芯片测序结果分析这个问题,去除芯片数据质量控制(这部分其实相当复杂),接下来就是差异基因筛选和基因功能注释分析了。
基因功能注释
属于芯片分析流程中最末端的生物学解读部分,相当于是临门一脚吧。这部分也是整个分析流程中最为灵活的部分,虽然它也有自身的一些套路。
吐槽一下现在文章中非常套路的热图
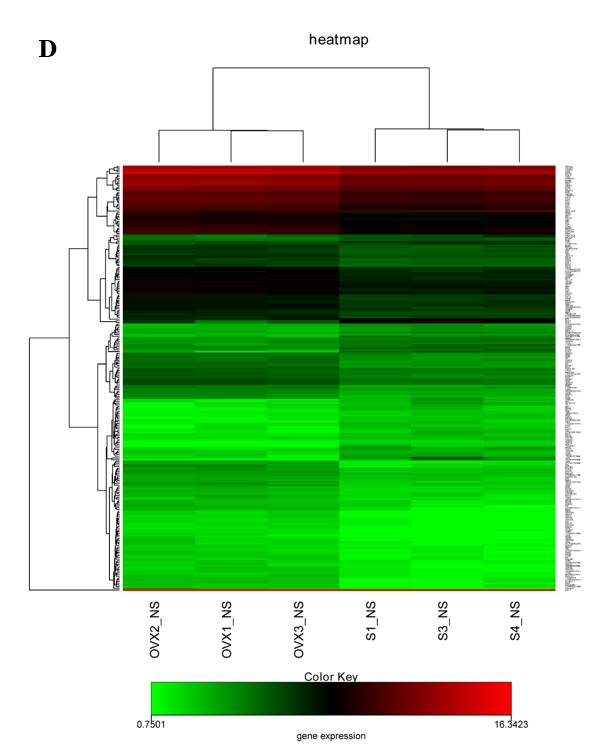
实验设计非常简单,2组,3vs3,差异表达基因的定义很明了,不是上调就是下调,通过阈值筛选以后,图形展示结果是早可以预见的,毫无意外。既然这样,为什么不直接列个表?回到热图的初衷,这是一个
聚类分析
,目的是找出表达轮廓相近基因,以此来推断它们在功能上存在关联。如果说一张热图仅仅是为了说明差异基因分的很开,筛选标准是OK的,我的实验分组是OK的,那么称它为一张“田”字红绿色盲测试图并不为过。
基因功能分析用GO和KEGG这一套,简单地罗列数据就OK了。
不是说套路有什么错,婴儿学说话,刚学的时候他也是不知道其中的含义的,但是仅仅停留在套路上,还是走不远的。
基因功能注释的过程是先聚类再检索功能,还是先检索功能再聚类?聚类的时候采用什么样的标准?生物功能富集检验的时候采用什么样的算法?这些都是大有可为的地方。
吐槽了这么多,最后给大家科普一下
生物功能富集检验
的方法。
在文章Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges中,作者按照方法论的不同,将功能富集分析分为三代:
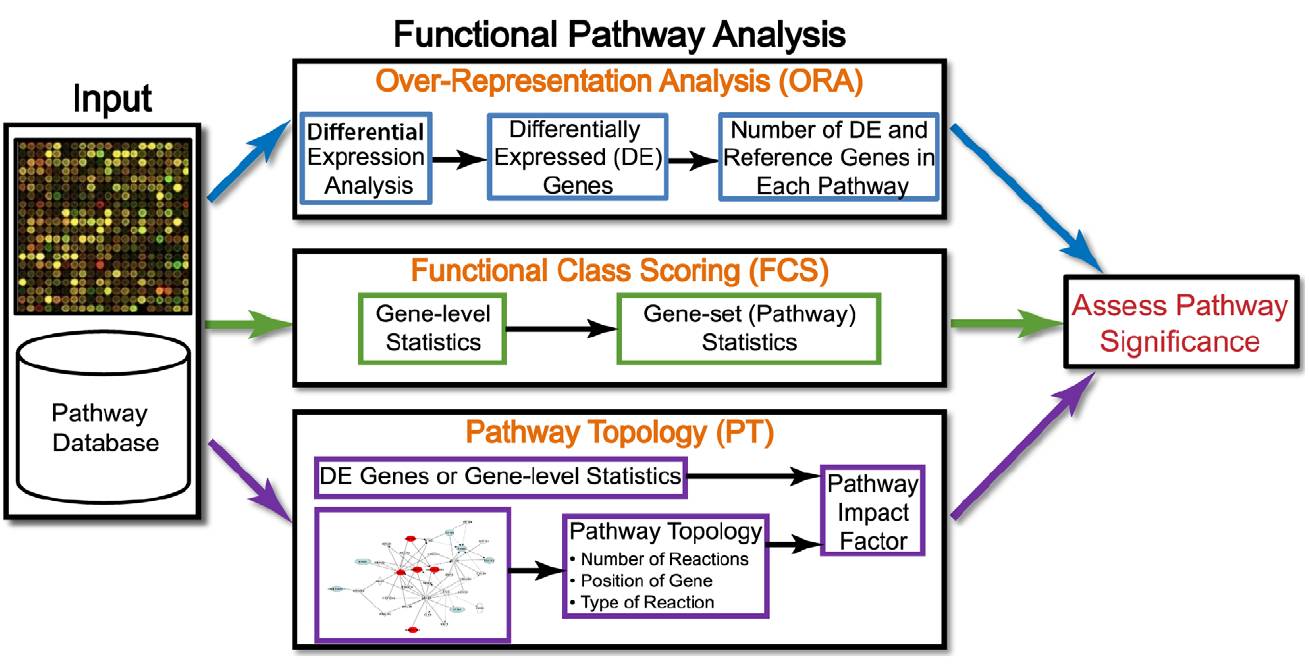
这三种方法本质上的区别在于
输入数据的形式
1、
ORA
:这是我们最常用的一种方法,这类方法以
Fisher's exact test
为代表。这类方法最为简单, 它只需要输入一个由差异基因构成的列表,这个列表中所有的基因都是平等的,没有权重或者顺序。
2、
FCS
:这类方法以
GSEA
为代表,小张之前也介绍过
GSEA是个什么鬼(上)?
,
GSEA是个什么鬼?(下)
。它不光需要输入给定基因列表,而且需要赋予每个基因一个感兴趣的统计量,一般来讲,那就是t统计量或者Fold Change之类,整个的输入,是个排序列表。
3、
PT
:这类方法以
SPIA
为代表。它的输入需要在第一代或第二代方法的基础上,结合实际的生物信号通路的拓扑结构,就是需要提供例如,C基因在A和B基因上游,激活A,且抑制B,这类信息。这类方法非常理想,但是存在很多问题,比如pathway的数据库尚未完善,生物系统的pathway是在不同条件下是不断变化的,有点像是你站在上帝视角录入数据,所以这类方法是“政治上正确”的,目前还无法大规模实现。
那么ORA和FCS两种方法的优劣呢?
简单的说,ORA所用的方法Fisher 's exact test的优点是“
非常简单
”——操作简单;缺点是“
非常简单
”——它需要回答“一个基因是否是差异表达”的问题,且答案只能是,Yes or No,而这个差异基因的定义,取决于人为的
Cutoff
,人为因素太大。
FCS的代表作GSEA就稍微复杂一些了。
它的优点是能分析出一些并不具有显著差异表达的基因的生物学功能。
比如当一个转录因子被抑制之后,它的靶基因可能只有寥寥几个下调显著,其它都是微微下调,这种情况下,Fisher 's exact test就不能起作用了,这些微微下调的基因会被忽略。在The Functional Consequences of Variation in Transcription Factor Binding这篇文章中还提供了相应的证据,表明上面提出的这种现象确实是存在的:

那么GSEA分析存在哪些缺陷呢?
1、
要求区别非表达与表达基因
。
GSEA 的期望输入是当前条件下表达基因的排序列表,如果混入并未表达的基因,则会降低统计检验势。然而复杂的是,表达基因的检出是平台特异的,在这一层面来说的话,RNA-seq的数据更具优势,因为表达了就是表达了,没表达就是没表达,很简单。
2、
只对一侧富集的趋势敏感。
这是算法本身的特性导致的,在应用到生物数据上时,则需要引起额外的注意:许多生物学过程的关键基因对下游,可能既有激活作用,亦有抑制作用。这在转录因子中,尤为常见。比如下面这个NF-kB的富集结果,左侧的趋势较右侧的弱一些,可能就会漏检。所以做GSEA的时候,做一次两端富集,再做一次单端富集可以比较好的避免这样的问题。
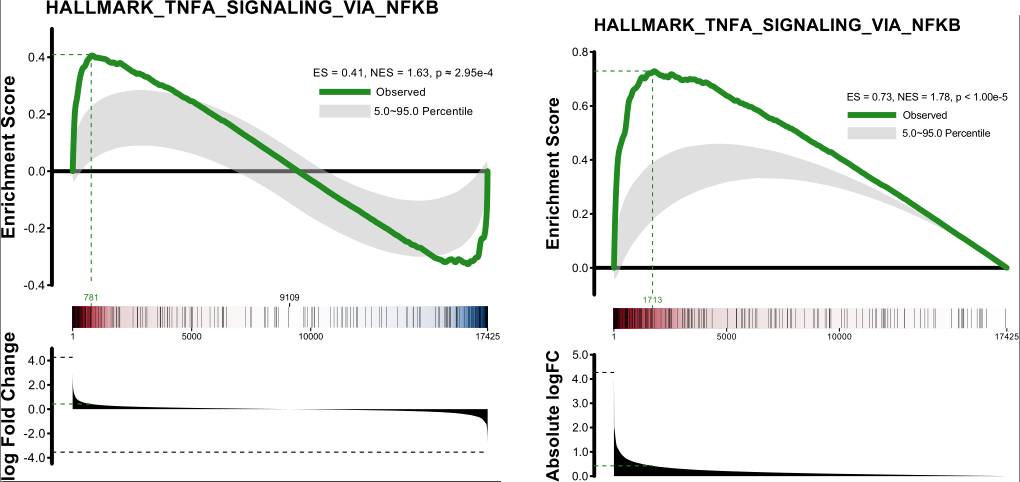
好了,今天扯这么多其实就是想跟大家谈谈对生信的一些看法,生信的套路其实并不难,但是想要更进一步,还是需要付出比较多的努力的,并没有什么事可以随随便便可以成功的。
我之所以要做生物信息学分析是因为我想通过严谨的科学方法得到一些可靠的结论,而不是出于其它的什么目的。
长按二维码识别关注“小张聊科研”

关注后获取《科研修炼手册》1、2、3、4、5,基金篇精华合集





