题记:这篇读书笔记我们将继续深入研究分类回归技术:回归树、分类树、随机森林与极限梯度提升。本文参考Cory Lesmeister博士主编《Mastering Machine Learning with R》(第2版)一书。数据来自R语言MASS包自带biopsy,Pima.tr和Pima.te数据集,ElemStatLearn包自带prostate数据集。代码来自Cory Lesmeister博士主编《Mastering Machine Learning with R》这本书配套的代码,笔者对部分代码进行修改。本文是个人的读书笔记,仅用于学习交流使用,我只是一个知识的搬运工。
1. 背景知识
“最好的分类器可能是随机森林(RF),而最好的版本(用R开发,通过caret包调用)可以在84.3%的数据集上达到94.1%的最大正确率,超过了90%的其他分类器。” -- 费尔南德斯.德尔加多 等(2014)
上文引自费尔南德斯德尔加多等人在《机器学习研究期刊》上发表的一篇论文,从这段话可以看出,我们在这一章要讨论的技术有多么强大,特别是用于解决分类问题时。当然,它们不是永恒的最优解,但确实提供了一个非常好的起点。
我们之前研究的技术或用于预测定量结果,或用于预测分类问题。本章要讨论的技术对这两类问题都适用,但处理问题的方式和前几章不同。本章我们并不引入新的问题,而是把这些技术用于前面已经解决的问题,着眼于这些技术能否提高预测能力。总而言之,本章案例的目的就是要看看是否能继续改善以前的模型。
第一个要讨论的项目是基础的决策树,模型建立和解释都非常简单。但决策树模型的表现并不如我们研究过的模型那样好,比如支持向量机模型;也比不上我们将要学习的模型,比如神经网络模型。所以,我们要讨论的是建立多个(有时是上百个)不同的树,然后把每个树的结果组合在一起,最后形成一个综合的预测。
正像本章开头引用的论文所说,这些方法产生的效果不次于甚至超过书中涉及的所有其他技术。这些技术被称为随机森林和梯度提升树。此外,在讨论案例之余,我们还会介绍如何在数据集上应用随机森林方法,以帮助实现特征消除或较特征选择。
2. 回归树
要想理解基于树的方法,比较容易的方式是从预测定量结果开始,然后学习它是如何解决分类问题的。树方法的精髓就是划分特征,从第一次分裂开始就要考虑如何最大程度改善RSS,然后持续进行“树杈”分裂,直到树结束。后面的划分并不作用于全体数据集,而仅作用于上次划分时落到这个分支之下的那部分数据。这个自顶向下的过程被称为“递归划分”。这个过程是贪婪的,这个名词在研究机器学习方法时会经常遇到。贪婪的含义是指算法在每次分裂中都追求最大程度减少RSS,而不管以后的划分中表现如何。这样做可能会生成一个带有无效分支的树,尽管偏差很小,但是方差很大。为了避免这个问题,生成完整的树之后,你要对树进行剪枝,得到最优的解。
这种方法的优点是可以处理高度非线性关系,但它还存在一些潜在的问题:一个观测被赋予所属终端节点的平均值,这会损害整体预测效果(高偏差)。相反,如果你一直对数据进行划分,树的层次越来越深,这样可以达到低偏差的效果,但是高方差又成了问题。和其他方法一样,你也可以用交叉验证来选择合适的深度。
3. 分类树
分类树与回归树的运行原理是一样的,区别在于决定分裂过程的不是RSS,而是误差率。这里的误差率不是你想的那样,简单地由误分类的观测数除以总观测数算出。实际上,进行树分裂时,误分类率本身可能会导致这样一种情况:你可以从下次分裂中获得一些有用信息,但误分类率却没有改善。我们看一个例子。
假设有一个节点N0,节点中有7个标号为N0的观测和3个标号为Yes的观测,我们就可以说误分类率为30%。记住这个数,然后通过另一种误差测量方式进行计算,这种方式称为基尼指数。单个节点的基尼指数计算公式如下:
基尼指数 = 1 - (类别1的概率)^2 - (类别2的概率)^2
所以,对于N0,基尼指数为1 - (0.7)^2 - (0.3)^2,等于0.42,与之相对的误分类率为30%。
将这个例子讨论得更深入一些,假设将节点N0分裂成两个节点N1和N2,N1中有3个观测属于类别1,没有属于类别2的观测;N2中有4个观测属于类别1,3个属于类别2。现在,树的这个分支的整体的误分类率还是30%,那么看看整体的基尼指数是如何改善的:
(1)基尼指数(N1) = 1 - (3/3)^2 - (0/3)^2 = 0
(2)基尼指数(N2) = 1 - (4/7)^2 - (3/7)^2 = 0.49
(3)新基尼指数 = (N1比例×基尼指数(N1))+(N2比例×基尼指数(N2)) = (0.3×0) + (0.7×0.49) = 0.343
通过一次基于替代误差率的分裂,我们确实改善了模型的不纯度,将其从原来的0.42减小到0.343,误分类率却没有变化。rpart()包就是使用Gini指数测量误差的,在本章中我们会使用这个程序包。
4. 随机森林
为了显著提高模型的预测能力,我们可以生成多个树,然后将这些树的结果组合起来。随机森林技术在模型构建过程中使用两种奇妙的方法,以实现这个构想。第一个方法称为自助聚集,或称装袋。
在装袋法中,使用数据集的一次随机抽样建立一个独立树,抽样的数量大概为全部观测的2/3(请记住,剩下的1/3被称为袋外数据,out-of-bag)。这个过程重复几十次或上百次,最后取平均结果。其中每个树都任其生长,不进行任何基于误差测量的剪枝,这意味着每个独立树的方差都很大。但是,通过对结果的平均化处理可以降低方差,同时又不增加偏差。
另一个在随机森林中使用的方法是,对数据进行随机抽样(装袋)的同时,独立树每次分裂时对输入特征也进行随机抽样。在randomForest包中,我们使用随机抽样数的默认值来对预测特征进行抽样。对于分类问题,默认值为所有预测特征数量的平方根;对于回归问题,默认值为所有预测特征数量除以3。在模型调优过程中,每次树分裂时,算法随机选择的预测特征数量是可变的。
通过每次分裂时对特征的随机抽样以及由此形成的一套方法,你可以减轻高度相关的预测特征的影响,这种预测特征在由装袋法生成的独立树中往往起主要作用。这种方法还使你不必思索如何减少装袋导致的高方差。独立树彼此之间的相关性减少之后,对结果的平均化可以使泛化效果更好,对于异常值的影响也更加不敏感,比仅进行装袋的效果要好。
5. 梯度提升
梯度提升法的主要思想是,先建立一个某种形式的初始模型(线性、样条、树或其他),称为基学习器;然后检查残差,在残差的基础上围绕损失函数拟合模型。损失函数测量模型和现实之间的差别,例如,在回归问题中可以用误差的平方。一直继续这个过程,直到满足某个特定的结束条件。这与下面的情形有点相似:一个学生进行模拟考试,100道题中错了30道,然后只研究那30道错题;在下次模考中,30道题中又错了10道,然后只研究那10道题,以此类推。如果你想更加深入地研究背后的理论,可以参考Frontiers in Neurorobotics, Gradient boosting machines,a tutorial, Natekin A., Knoll A. (2013)(http://www.ncbi.nlm.nih.gov/pmc/atricles/PMC3885826/).
刚才提到过,梯度提升方法可以用于很多不同类型的基分类器,本章只集中讨论基于树的学习这个主题。每个树迭代的次数都很少,我们要通过一个调优参数决定树的分裂次数,这个参数称为交互深度。事实上,有些树很小,只可以分裂一次,这样的树就被称为“树桩”。这些树依次按照损失函数拟合残差,直至达到我们指定的树的数量(我们的结束条件)。使用Xgboost包进行建模的过程中,有一些参数需要调整。Xgboost表示eXtreme Gradient Boosting。这个程序包表现优异,广泛应用于网上的数据科学竞赛。在下面这个网站中,你可以找到关于提升树和Xgboost的非常详尽的背景资料:http://xgboost.readthedocs.io/en/latest/model.html.
在下面的案例中,我们要看看如何找到最优的超参数,并使模型生成有意义的结果和预测。这些参数之间会互相作用,如果你只攻一点而不顾其他,模型的效果可能会越来越差。在调参过程中,caret包会助你一臂之力。
6. 案例分析
在前几章中,我们通过努力建立了一些模型,本章的总体目标是看看我们能否提高这些模型的预测能力。对于回归问题,重新回到第4章中的前列腺癌数据集,以0.444的均方误差作为基准,看看能否改善。对于分类问题,我们使用第3章中的乳腺癌数据集和第5章中的皮玛印第安人糖尿病数据集。在乳腺癌数据集中,我们取得了97.6%的预测正确率。在糖尿病数据集中,正确率是79.6%,我们希望再提高一些。随机森林和提升方法都会用于这3个数据集,简单树模型只用于乳腺癌数据集和前列腺癌数据集。
7. 模型构建与模型评价
要进行模型构建,需要加载7个不同的R包。我们会实验每种技术,并与前面章节中介绍的方法进行对比,看看它们在数据分析中会有什么突出表现。
(1)回归树 我们直接跳到前列腺癌数据集,但首先要加载所需的R包。当然,要确定在加载之前你已经安装了它们:
library(rpart) #classification and regression trees
library(partykit) #treeplots
library(MASS) #breast and pima indian data
library(ElemStatLearn) #prostate data
library
(randomForest) #random forests
library(xgboost) #gradient boosting
library(caret) #tune hyper-parameters
先用prostate数据集做回归。使用ifelse()函数将gleason评分编码为指标变量,划分训练数据集和测试数据集,训练数据集为pros.train,测试数据集为pros.test,如下所示:
data(prostate)
prostate$gleason 6, 0, 1)
pros.train TRUE)[, 1:9]
pros.test = subset(prostate, train == FALSE)[, 1:9]
要在训练数据集上建立回归树,需要使用party包中的rpart()函数,语法与我们在其他建模技术中使用过的非常类似:
set.seed(123)
tree.pros
可以调出这个对象,然后检查每次分裂的误差,以决定最优的树分裂次数:
tree.pros$cptable
## CP nsplit rel error xerror xstd
## 1 0.35852251 0 1.0000000 1.0364016 0.1822698
## 2 0.12295687 1 0.6414775 0.8395071 0.1214181
## 3 0.11639953 2 0.5185206 0.7255295 0.1015424
## 4 0.05350873 3 0.4021211 0.7608289 0.1109777
## 5 0.01032838 4 0.3486124 0.6911426 0.1061507
## 6 0.01000000 5 0.3382840 0.7102030 0.1093327
这个表对于我们的分析非常重要。标号为CP的第一列是成本复杂性参数,第二列nsplit是树分裂的次数,rel error列表示相对误差,即某次分裂的RSS除以不分裂的RSS(RSS(k)/RSS(0)),xerror和xstd都是基于10折交叉验证的,xerror是平均误差,xstd是交叉验证过程的标准差。可以看出,5次分裂在整个数据集上产生的误差最小,但使用交叉验证时,4次分裂产生的误差略微更小。可以使用plotcp()函数查看统计图:
plotcp(tree.pros)
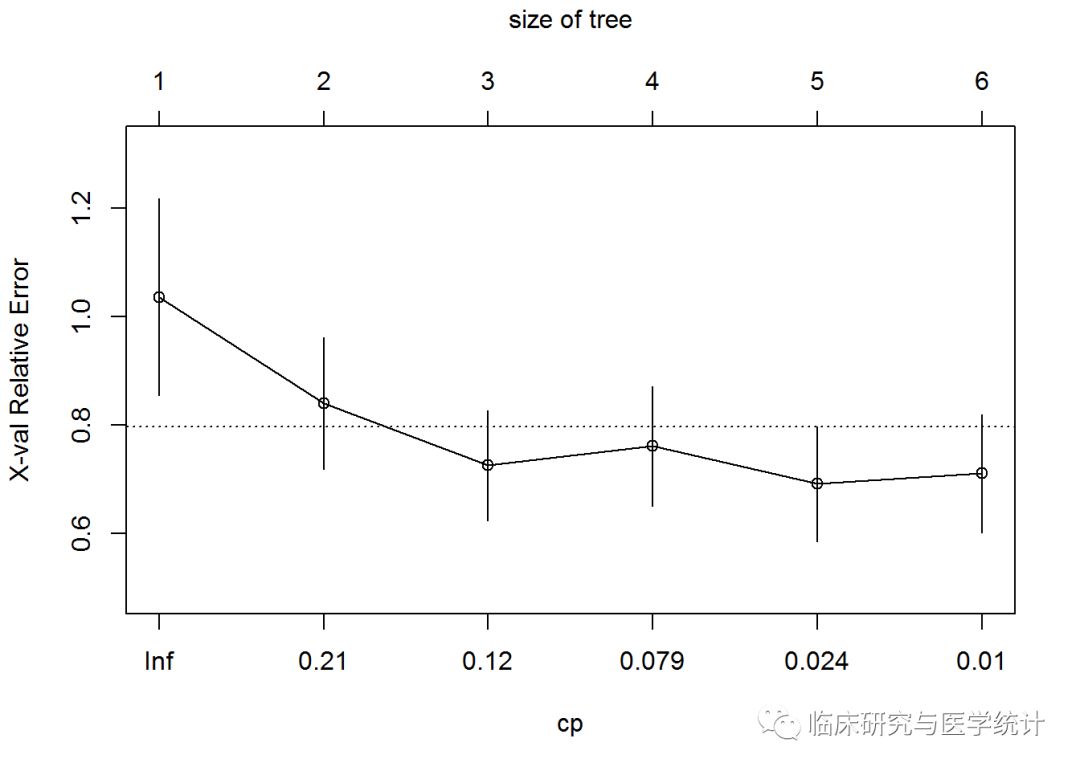
这幅图使用误差条表示树的规模和相对误差之间的关系,误差条和树规模是对应的。选择树的规模为5,也就是经过4次分裂可以建立一个新的树对象,这个树的xerror可以通过剪枝达到最小化。剪枝的方法是先建立一个cp对象,将这个对象和表中第5行相关联,然后使用prune()函数完成剩下的工作:
cp 5, ])
prune.tree.pros
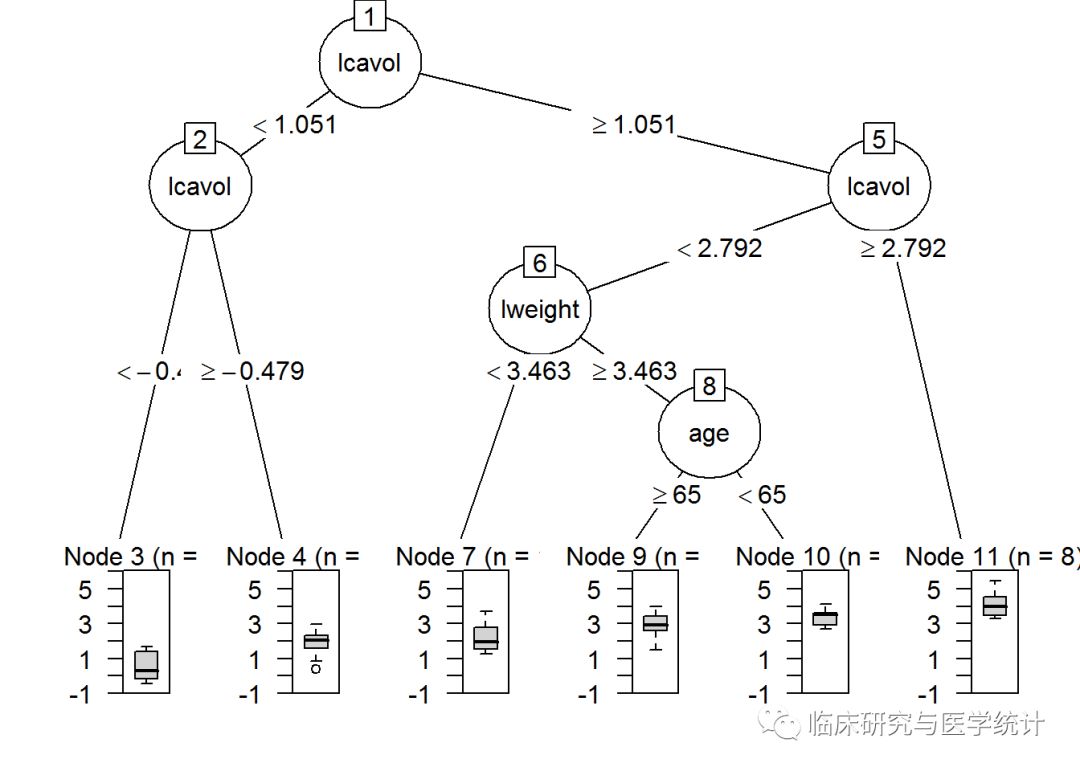
完成上面的代码后,可以用统计图比较完整树和剪枝树。由partykit包生成的树图明显优于party包生成的,在plot()函数中,你可以简单地使用as.party()函数作为包装器函数:
plot(as.party(tree.pros))
同样地,使用as.party()函数处理剪枝树:
plot(as.party(prune.tree.pros))
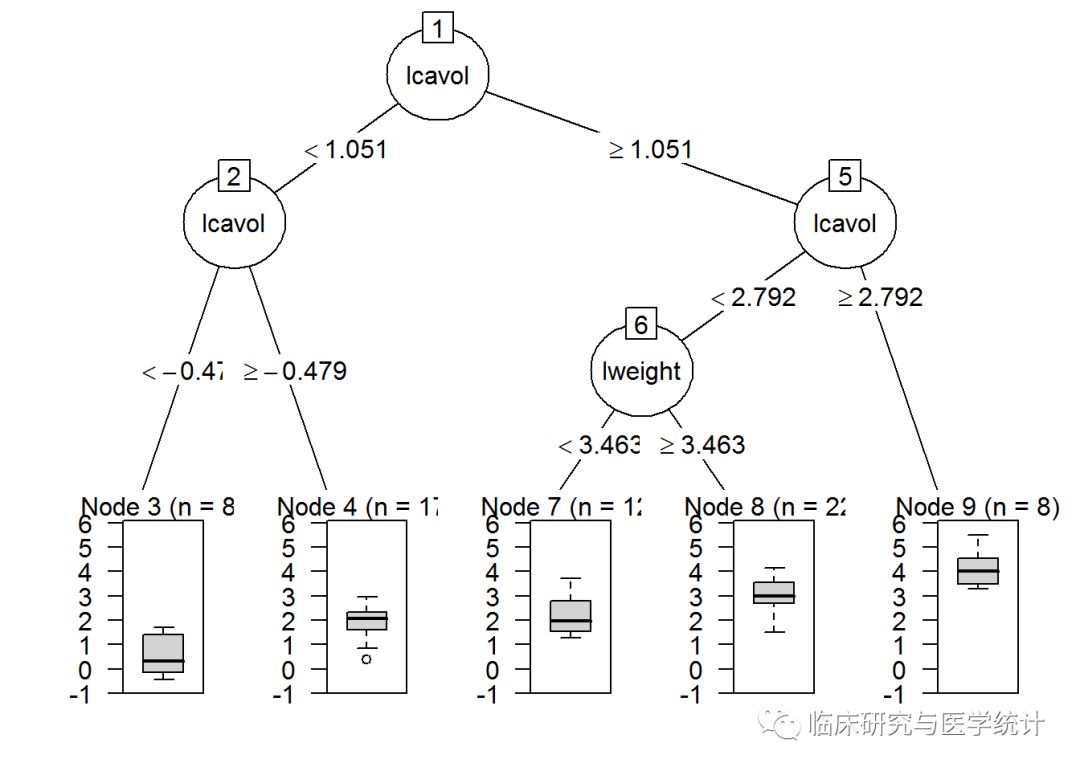
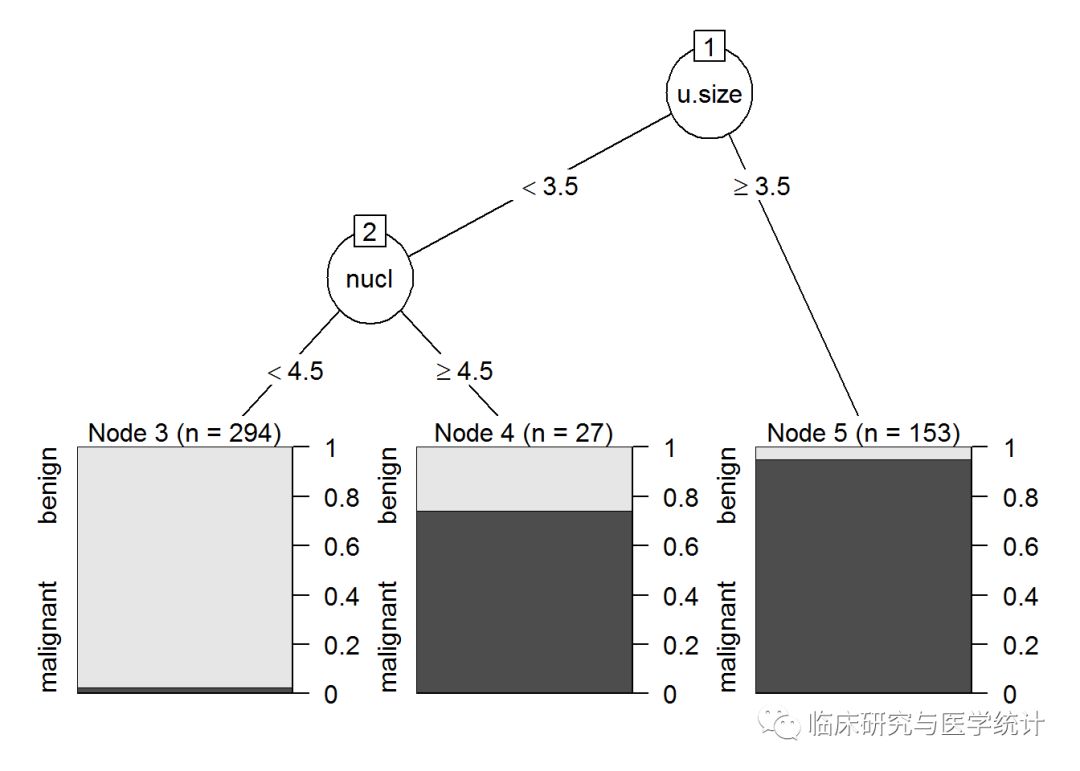
请注意,除了最后一次分裂(完整树包含变量age),两个树是完全一样的。有趣的是,树的第一次分裂和第二次分裂都与肿瘤大小的对数(lcavol)有关。这两张图的信息量都很大,因为它们显示了树的分裂、节点、每节点观测数,以及预测结果的箱线图。
下面我们看看剪枝树在测试集上表现如何。在测试数据上使用predict()函数进行预测,并建立一个对象保存这些预测值。然后用预测值减去实际值,得到误差,最后算出误差平方的平均值:
party.pros.test #calculate residualmean(rpart.resid^2)
## [1] 0.5267748
第4章中的基准MSE为0.44,我们的努力并没有改善预测结果。但是,这种方法并非一无是处。从生成的树图中可以看出对响应变量影响最大的特征,并且很容易解释。就像引言中说过的,树模型很容易理解和解释,这往往比正确率重要得多。
(2)分类树 对于分类问题,我们先像第3章一样准备好乳腺癌数据。加载数据之后,你要删除患者ID,对特征进行重新命名,删除一些缺失值,然后建立训练数据集和测试数据集。如下所示:
data(biopsy)
biopsy 1]
names(biopsy) "thick", "u.size"
, "u.shape", "adhsn", "s.size", "nucl", "chrom", "n.nuc", "mit", "class")
biopsy.v2 123) #random number generator
ind 2, nrow(biopsy.v2), replace = TRUE, prob = c(0.7, 0.3))
biop.train 1, ] #the training data set
biop.test 2, ] #the test data set
准备好合适的数据之后,使用和前面回归问题一样的代码风格来解决分类问题。建立分类树之前,要确保结果变量是一个因子,可以用str()函数进行检查:
str(biop.test)
## 'data.frame': 209 obs. of 10 variables:
## $ thick : int 5 6 4 2 1 7 6 7 1 3 ...
## $ u.size : int 4 8 1 1 1 4 1 3 1 2 ...
## $ u.shape: int 4 8 1 2 1 6 1 2 1 1 ...
## $ adhsn : int 5 1 3 1 1 4 1 10 1 1 ...
## $ s.size : int 7 3 2 2 1 6 2 5 2 1 ...
## $ nucl : int 10 4 1 1 1 1 1 10 1 1 ...
## $ chrom : int 3 3 3 3 3 4 3 5 3 2 ...
## $ n.nuc : int 2 7 1 1 1 3 1 4 1 1 ...
## $ mit : int 1 1 1 1 1 1 1 4 1 1 ...
## $ class : Factor w/ 2 levels "benign","malignant": 1 1 1 1 1 2 1 2 1 1 ...
## - attr(*, "na.action")= 'omit' Named int 24 41 140 146 159 165 236 250 276 293 ...
## ..- attr(*, "names")= chr "24" "41" "140" "146" ...
先生成树,然后检查输出中的表格,找到最优分裂次数:
set.seed(123)
tree.biop
## CP nsplit rel error xerror xstd
## 1 0.79651163 0 1.0000000 1.0000000 0.06086254
## 2 0.07558140 1 0.2034884 0.2674419 0.03746996
## 3 0.01162791 2 0.1279070 0.1453488 0.02829278
## 4 0.01000000 3 0.1162791 0.1744186 0.03082013
交叉验证误差仅在两次分裂后就达到了最小值(见第3行)。现在可以对树进行剪枝,再在图中绘制剪枝树,看看它在测试集上的表现:
cp 3, ])
prune.tree.biop = prune(tree.biop, cp
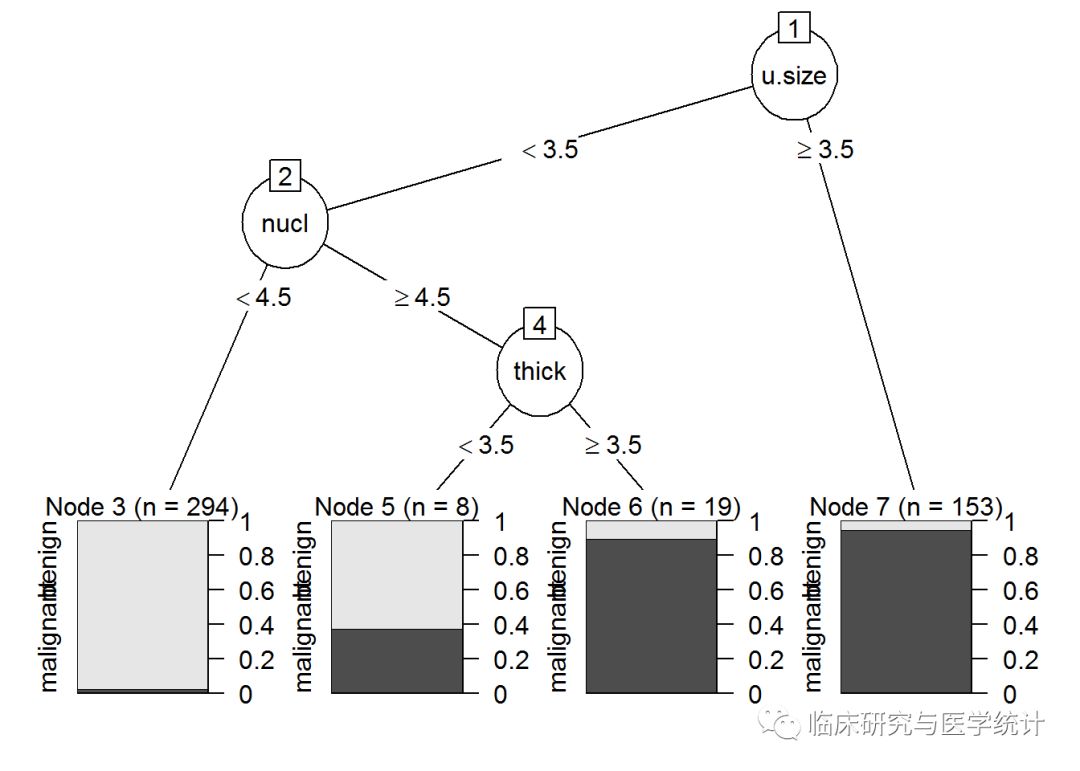
plot(as.party(prune.tree.biop))
从对树图的检查中可以发现,细胞大小均匀度是第一个分裂点,第二个是nuclei。完整树还有一个分支是细胞浓度。使用predict()函数并指定type=“class”,在测试集上进行预测。如下所示:
rparty.test "class")
table(rparty.test, biop.test$class)
## rparty.test benign malignant
## benign 136 3
## malignant 6 64
(136+64)/209
## [1] 0.9569378
只有两个分支的基本树模型给出了差不多96%的正确率。这虽然比不上Logistic回归的正确率97.6%,但足以激励我们相信,在随后的随机森林方法中会得到更好的改善效果。
(3)随机森林回归 在这一节中,我们还是首先处理前列腺癌数据集,然后处理乳腺癌数据集和皮玛印第安人糖尿病数据集,使用的程序包是randomForest。建立一个随机森林对象的通用语法是使用randomForest()函数,指定模型公式和数据集这两个基本参数。回想一下每次树迭代默认的变量抽样数,对于回归问题,是p/3;对于分类问题,是p的平方根,p为数据集中预测变量的个数。对于大规模数据集,就p而言,你可以调整mtry参数,它可以确定每次迭代的变量抽样数值。如果p小于10,可以省略上面的调整过程。想在多特征数据集中优化mtry参数时,可以使用caret包,或使用randomForest包中的tuneRF()函数。了解这些情况之后,即可建立随机森林模型并检查模型结果。如下所示:
set.seed(123)
rf.pros
## Call:
## randomForest(formula = lpsa ~ ., data = pros.train)
## Type of random forest: regression
## Number of trees: 500
## No. of variables tried at each split: 2
## Mean of squared residuals: 0.6936697
## % Var explained: 51.73
调用rf.pros对象,可以知道随机森林生成了500个不同的树(默认设置),并且在每次树分裂时随机抽出两个变量。结果的MSE为0.68,差不多53%的方差得到了解释。下面看看能否对默认数量的树做一些改善。过多的树会导致过拟合,当然,“多大的数量是‘过多’”依赖于数据规模。两种方式可以帮助我们达到目的,第一是做出rf.pros的统计图,另一件是求出最小的MSE:
plot(rf.pros)
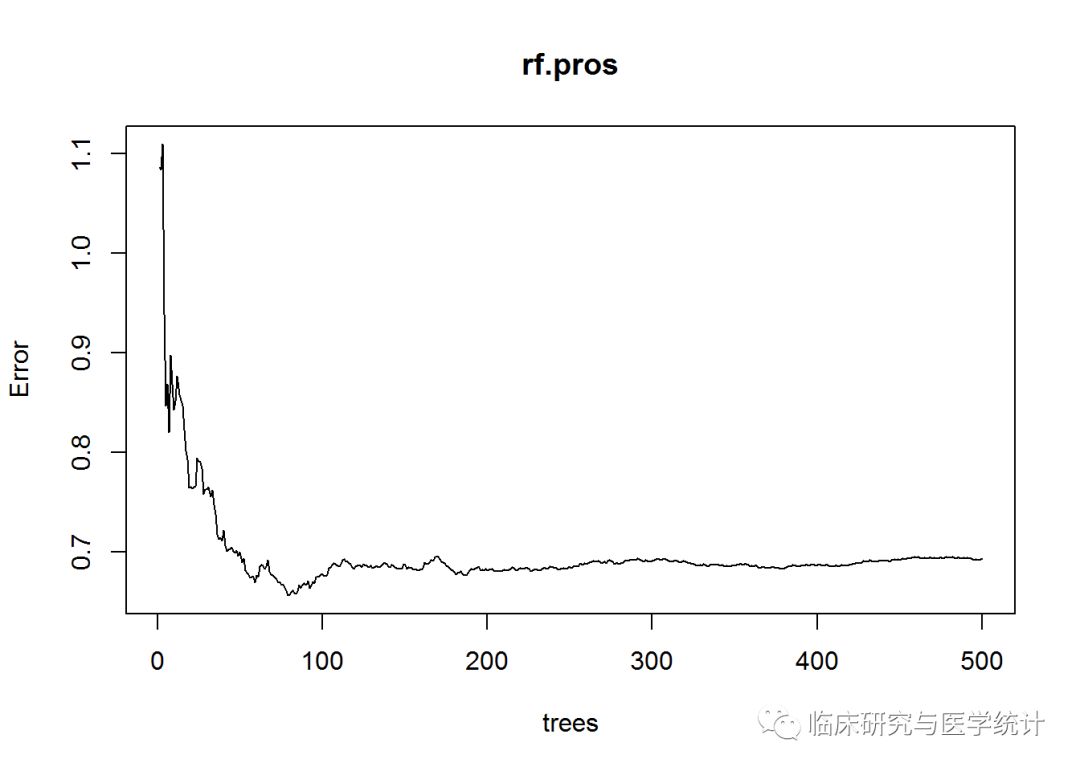
这个图表示MSE与模型中树的数量之间的关系。可以看出,树的数量增加时,一开始MSE会有显著改善,当森林中大约建立了100棵树之后,改善几乎停滞。可以通过which.min()函数找出具体的最优树数量,如下所示:
which.min(rf.pros$mse)
## [1] 80
可以试试有75棵树的随机森林,在模型语法中指定ntree = 75即可:
set.seed(123)
rf.pros.2 75)
rf.pros.2
## Call:
## randomForest(formula = lpsa ~ ., data = pros.train, ntree = 75)
## Type of random forest: regression
## Number of trees: 75
## No. of variables tried at each split: 2
## Mean of squared residuals: 0.6672276
## % Var explained: 53.57
可以看到,MSE和解释方差都有一点微小的改善。对模型进行检验之前,先看看另一张统计图。如果使用自助重抽样和两个随机预测变量建立了75棵不同的树,要想将树的结果组合起来,需要一种方法确定哪些变量驱动着结果。独木不成林,你需要做出变量重要性统计图及相应的列表。Y轴是按重要性降序排列的变量列表,X轴是MSE改善百分比。请注意,在分类问题中,X轴应该是基尼指数的改善。使用varImpPlot()函数生成统计图:
varImpPlot(rf.pros.2, scale = TRUE,
main = "Variable Importance Plot - PSA Score")
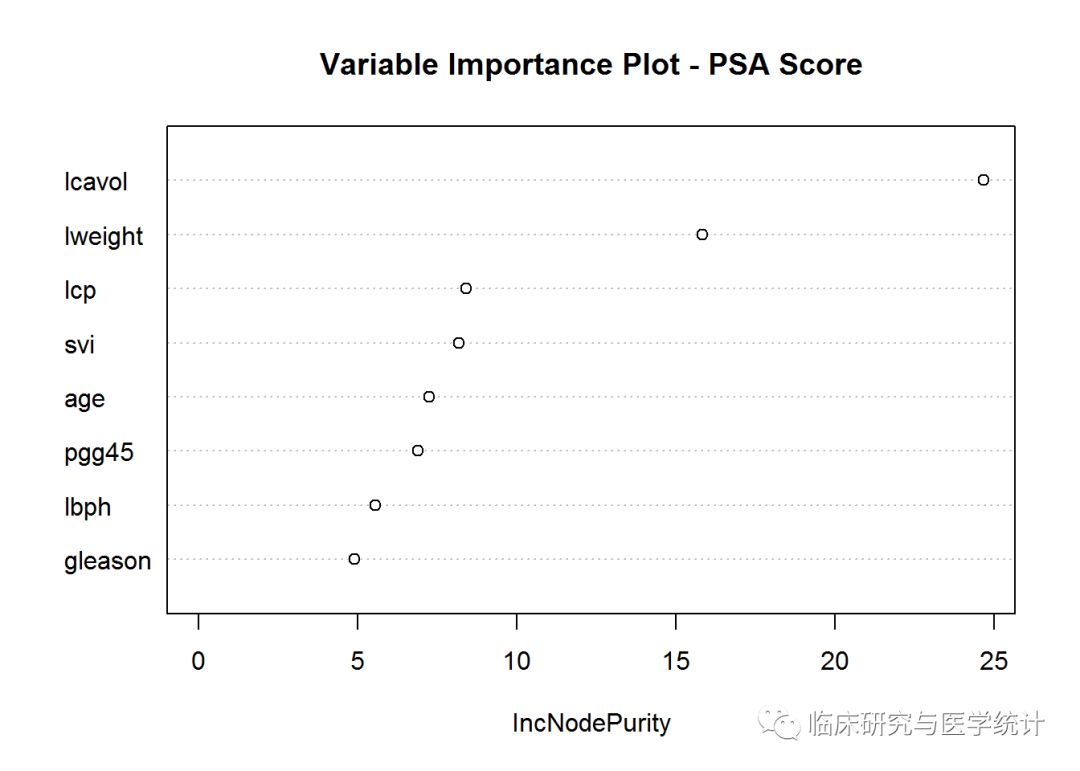
和单个树模型一致,lcavol是最重要的变量,lweight次之。如果想查看具体数据,可以使用importance()函数。如下所示:
importance(rf.pros.2)
## IncNodePurity
## lcavol 24.657359
## lweight 15.817242
## age 7.240911
## lbph 5.550491
## svi 8.175192
## lcp 8.387174
## gleason 4.879600
## pgg45 6.876621
现在可以看看模型在测试数据上的表现:
rf.pros.test #plot(rf.pros.test, pros.test$lpsa)
rf.resid #calculate residual
mean(rf.resid^2)
## [1] 0.5568805
MSE依然高于我们在第4章中使用LASSO得到的0.44,但也不比单个树模型更好。
(4)随机森林分类 你可能会对随机森林回归模型的表现感到失望,但这种技术的真正威力在于解决分类问题。我们从乳腺癌诊断数据开始,这个过程和在回归问题中的做法几乎一样:
set.seed(123)
rf.biop
## Call:
## randomForest(formula = class ~ ., data = biop.train)
## Type of random forest: classification
## Number of trees: 500
## No. of variables tried at each split: 3
## OOB estimate of error rate: 3.38%
## Confusion matrix:
## benign malignant class.error
## benign 294 8 0.02649007
## malignant 8 164 0.04651163
OOB(袋外数据)误差率为3.38%。随机森林还是由默认的全部500棵树组成。画出表示误差和树的数量关系的统计图:
plot(rf.biop)
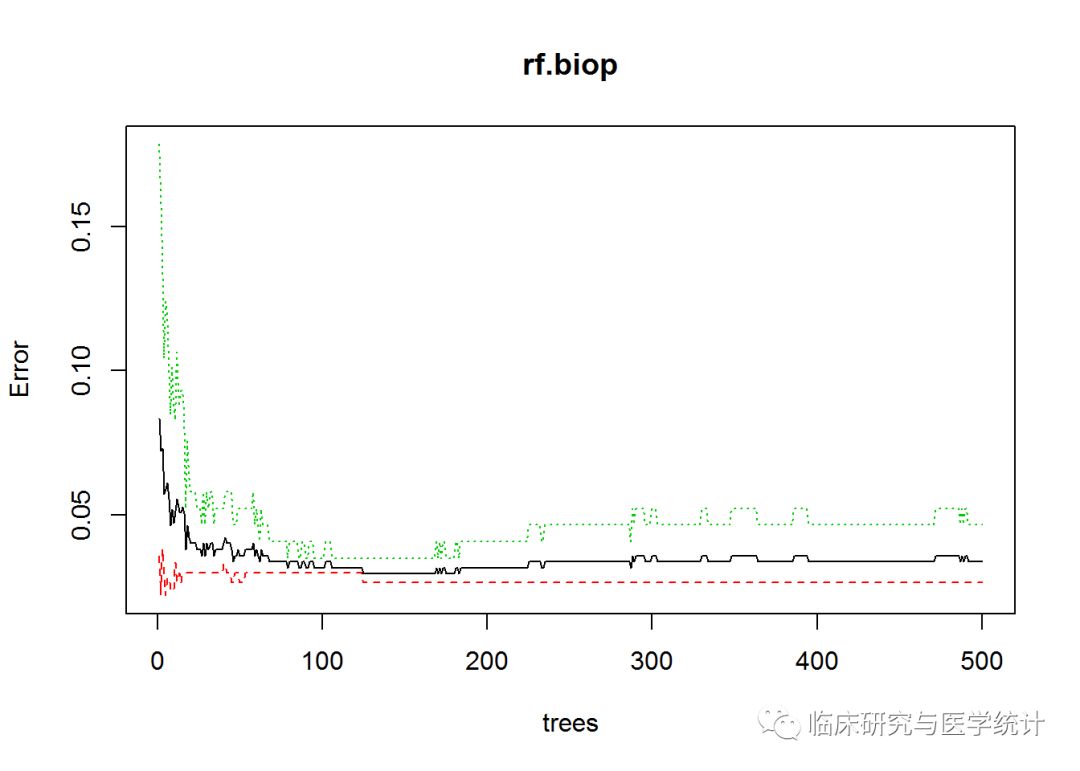
图中显示,误差和标准误差的最小值是在树的数量很少的时候取得的,可以使用which.min()函数找出具体值。和前面不同的一点是,需要指定第一列来得到误差率,这是整体误差率。算法会为每个类标号的误差率生成一个附加列,本例中不需要它们。模型结果中也没有mse,而是代之以err.rate。如下页代所示:
which.min(rf.biop$err.rate[, 1])
## [1] 125
只需19棵树就可以使模型正确率达到最优。实验一下,看看模型的表现:
set.seed(123
)
rf.biop.2 19)#getTree(rf.biop,1)
rf.biop.2
## Call:
## randomForest(formula = class ~ ., data = biop.train, ntree = 19)
## Type of random forest: classification
## Number of trees: 19
## No. of variables tried at each split: 3
## OOB estimate of error rate: 4.22%
## Confusion matrix:
## benign malignant class.error
## benign 293 9 0.02980132
## malignant 11 161 0.06395349
rf.biop.test "response")
table(rf.biop.test, biop.test$class)
## rf.biop.test benign malignant
## benign 138 0
## malignant 4 67
(139 + 67) / 209
## [1] 0.9856459
训练集上的误差率4.22%,在测试集上甚至表现得更好,误差率不到1.5%。测试集的209个观测中,只有3个观测被误分类,而且没有一个是误诊为“恶性”的。回忆一下,我们使用Logistic回归得到的正确率最好结果是97.6%,所以随机森林模型就是至今为止在乳腺癌数据上的最佳模型。进行下面的内容之前,先看看变量重要性统计图。
varImpPlot(rf.biop.2)
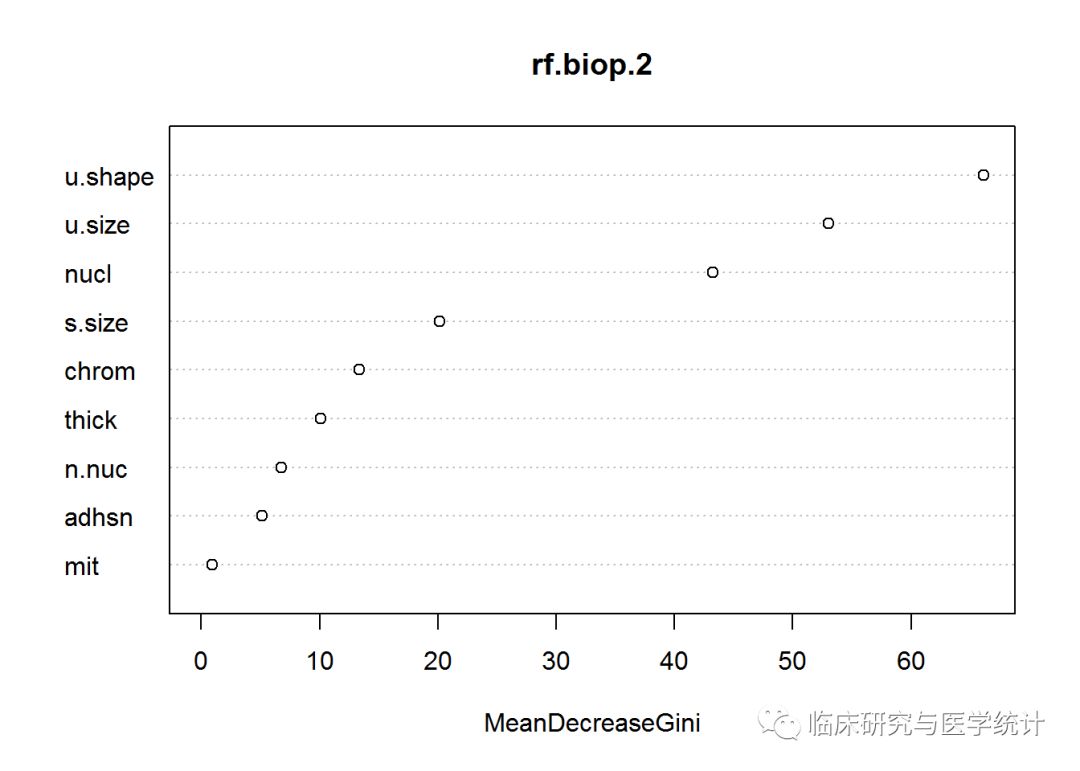
上图中,变量重要性是指每个变量对基尼指数平均减少量的贡献,此处的变量重要性与单个树分裂时有很大区别。回忆一下,单个树是在细胞大小均匀度开始分裂的(与随机森林一致),然后是nuclei,接着是细胞密度。这揭示了随机森林技术具有非常大的潜力,不但可以提高模型预测能力,还可以改善特征选择的结果。
现在,开始面对皮玛印第安人糖尿病模型的艰巨挑战。我们先做好数据准备,如下所示:
data(Pima.tr)
data(Pima.te)
pima 502)
ind 2, nrow(pima), replace = TRUE, prob = c(0.7, 0.3))
pima.train 1, ]
pima.test 2, ]
做完数据准备工作,下一步建立模型。如下所示:
set.seed(321)
rf.pima
## Call:
## randomForest(formula = type ~ ., data = pima.train)
## Type of random forest: classification
## Number of trees: 500
## No. of variables tried at each split: 2
##
## OOB estimate of error rate: 20.26%
## Confusion matrix:
## No Yes class.error
## No 235 27 0.1030534
## Yes 51 72 0.4146341
# plot(rf.pima)
我们在训练数据集上得到了20.26%的误分类误差率,并不比以前的结果更好。下面对树的数目进行优化,看看能否显著改善模型结果:
which.min(rf.pima$err.rate[,1])
## [1] 88
set.seed(321)
rf.pima.2 80)
rf.pima.2
## Call:
## randomForest(formula = type ~ ., data = pima.train, ntree = 80)
## Type of random forest: classification
## Number of trees: 80
## No. of variables tried at each split: 2
##
## OOB estimate of error rate: 21.04%
## Confusion matrix:
## No Yes class.error
## No 233 29 0.1106870
## Yes 52 71 0.4227642
当随机森林中有80棵树时,OOB误差并没有改善。随机森林能够在测试集上证明自己吗?我们来看一下。如下所示
rf.pima.test "response")
table(rf.pima.test, pima.test$type)
## rf.pima.test No Yes
## No 74 18
## Yes 19 36
(75
+33)/147
## [1] 0.7346939
#varImpPlot(rf.pima.2)
我们在测试集上只得到了大约73%的正确率,低于使用SVM的结果。虽然随机森林在糖尿病数据集上令人失望,但它已经证明了,自己在乳腺癌诊断方面是迄今为止最好的分类器。最后,我们实验一下梯度提升方法。
(5)极限梯度提升分类 我们提到过,在这一节要使用已经加载的xgboost包。因为这种方法的效果如雷贯耳,所以我们直接用它解决最有挑战性的皮玛印第安人糖尿病问题。正如在提升算法简介中所说,我们要调整一些参数。
(1)nrounds:最大迭代次数(最终模型中树的数量)。
(2)colsample_bytree:建立树时随机抽取的特征数量,用一个比率表示,默认值为1(使用100%的特征)。
(3)min_child_weight:对树进行提升时使用的最小权重,默认为1。
(4)eta:学习率,每棵树在最终解中的贡献,默认为0.3。
(5)gamma:在树中新增一个叶子分区时所需的最小减损。
(6)subsample:子样本数据占整个观测的比例,默认值为1(100%)。
(7)max_depth:单个树的最大深度。
使用expand.grid()函数可以建立实验网格,以运行caret包的训练过程。对于前面列出的参数,如果没有设定具体值,那么即使有默认值,运行函数时也会收到出错信息。下面的参数取值是基于我以前的一些训练迭代而设定的。建议各位亲自实验参数调整过程。使用以下命令建立网格:
grid = expand.grid(
nrounds = c(75, 100),
colsample_bytree = 1,
min_child_weight = 1,
eta = c(0.01, 0.1, 0.3), #0.3 is default,
gamma = c(0.5, 0.25),
subsample = 0.5,
max_depth = c(2, 3)
)
grid
## nrounds colsample_bytree min_child_weight eta gamma subsample
## 1 75 1 1 0.01 0.50 0.5
## 2 100 1 1 0.01 0.50 0.5
## 3 75 1 1 0.10 0.50 0.5
## 4 100 1 1 0.10 0.50 0.5
## 5 75 1 1 0.30 0.50 0.5
## 6 100 1 1 0.30 0.50 0.5
## 7 75 1 1 0.01 0.25 0.5
## 8 100 1 1 0.01 0.25 0.5
## 9 75 1 1 0.10 0.25 0.5
## 10 100 1 1 0.10 0.25 0.5
## 11 75 1 1 0.30 0.25 0.5
## 12 100 1 1 0.30 0.25 0.5
## 13 75 1 1 0.01 0.50 0.5
## 14 100 1 1 0.01 0.50 0.5
## 15 75 1 1 0.10 0.50 0.5
## 16 100 1 1 0.10 0.50 0.5
## 17 75 1 1 0.30 0.50 0.5
## 18 100 1 1 0.30 0.50 0.5
## 19 75 1 1 0.01 0.25 0.5
## 20 100 1 1 0.01 0.25 0.5
## 21 75 1 1 0.10 0.25 0.5
## 22 100 1 1 0.10 0.25 0.5
## 23 75 1 1 0.30 0.25 0.5
## 24 100 1 1 0.30 0.25 0.5
## max_depth
## 1 2
## 2 2
## 3 2
## 4 2
## 5 2
## 6 2
## 7 2
## 8 2
## 9 2
## 10 2
## 11 2
## 12 2
## 13 3
## 14 3
## 15 3
## 16 3
## 17 3
## 18 3
## 19 3
## 20 3
## 21 3
## 22 3
## 23 3
## 24 3
以上命令会建立一个具有24个模型的网格,caret包会运行这些模型,以确定最好的调优参数。此处必须提示各位,对于我们现在所用的这种规模的数据集,代码运行过程只需几秒钟,但对于一些大数据集,这个运行过程可能需要几小时。所以,在时间非常宝贵的情况下,你必须应用自己的判断力,并使用小数据样本来找出合适的调优参数,否则硬盘空间可能不足。
使用car包的train()函数之前,我要创建一个名为cntrl的对象,来设定trainControl的参数。这个对象会保存我们要使用的方法,以训练调优参数。我们使用5折交叉验证,如下所示:
cntrl = trainControl(
method = "cv",
number = 5,
verboseIter = TRUE,
returnData = FALSE,
returnResamp = "final" )
要使用train.xgb()函数,只需和训练其他模型一样,设定好所需参数即可:训练数据集、标号、训练控制对象和实验网格。记得要设定随机数种子:
set.seed(1)
train.xgb = train(
x = pima.train[, 1:7],
y = ,pima.train[, 8],
trControl = cntrl,
tuneGrid = grid,
method = "xgbTree")
## + Fold1: eta=0.01, max_depth=2, gamma=0.25, colsample_bytree=1, min_child_weight=1, subsample=0.5, nrounds=100
......
## - Fold5: eta=0.30, max_depth=3, gamma=0.50, colsample_bytree=1, min_child_weight=1, subsample=0.5, nrounds=100
## Aggregating results
## Selecting tuning parameters
## Fitting nrounds = 100, max_depth = 2, eta = 0.01, gamma = 0.5, colsample_bytree = 1, min_child_weight = 1, subsample = 0.5 on full training set
因为在trControl中设定了verboseIter为TURE,所以可以看到每折交叉验证中的每次训练迭代。
调用train.xgb这个对象可以得到最优的参数,以及每种参数设置的结果,如下所示(有所简省):
train.xgb
## eXtreme Gradient Boosting
## No pre-processing
## Resampling: Cross-Validated (5 fold)
## Summary of sample sizes: 308, 308, 308, 308, 308
## Resampling results across tuning parameters:
## eta max_depth gamma nrounds Accuracy Kappa
## 0.01 2 0.25 75 0.7974026 0.5016374
## 0.01 2 0.25 100 0.8000000 0.5086971
## 0.01 2 0.50 75 0.7974026 0.5011633
## 0.01 2 0.50 100 0.8025974 0.5175787
## 0.01 3 0.25 75 0.7922078 0.4922910
## 0.01 3 0.25 100 0.7974026 0.5066888
## 0.01 3 0.50 75 0.7948052 0.4997955
## 0.01 3 0.50 100 0.7948052 0.4997955
## 0.10 2 0.25 75 0.7870130 0.4863234
## 0.10 2 0.25 100 0.7896104 0.4946068
## 0.10 2 0.50 75 0.7974026 0.5208894
## 0.10 2 0.50 100 0.7922078 0.5078930
## 0.10 3 0.25 75 0.7870130 0.4871247
## 0.10 3 0.25 100 0.7870130 0.4946006
## 0.10 3 0.50 75 0.7896104 0.5000596
## 0.10 3 0.50 100 0.7948052 0.5131372
## 0.30 2 0.25 75 0.7688312 0.4604660
## 0.30 2 0.25 100 0.7688312 0.4599298
## 0.30 2 0.50 75 0.7818182 0.4853625
## 0.30 2 0.50 100 0.7636364 0.4481784
## 0.30 3 0.25 75 0.7974026 0.5112360
## 0.30 3 0.25 100 0.7740260 0.4589619
## 0.30 3 0.50 75 0.7818182 0.4946846
## 0.30 3 0.50 100 0.7792208 0.4877327
### Tuning parameter 'colsample_bytree' was held constant at a value of
## 1
## Tuning parameter 'min_child_weight' was held constant at a value of
## 1
## Tuning parameter 'subsample' was held constant at a value of 0.5
## Accuracy was used to select the optimal model using the largest value.
## The final values used for the model were nrounds = 100, max_depth = 2,
## eta = 0.01, gamma = 0.5, colsample_bytree = 1, min_child_weight = 1
## and subsample = 0.5.
由此可以得到最优的参数组合来建立模型。模型在训练数据上的正确率是81%,Kappa值是0.55。下面要做的事情有点复杂,但我认为这才是最佳实践。首先创建一个参数列表,供Xgboost包的训练函数xgb.train()使用。然后将数据框转换为一个输入特征矩阵,以及一个带标号的数值型结果列表(其中的值是0和1)。接着,将特征矩阵和标号列表组合成符合要求的输入,即一个xgb.Dmatrix对象。代码如下:
param "binary:logistic",
booster = "gbtree",
eval_metric = "error",
eta = 0.1,
max_depth = 2,
subsample = 0.5,
colsample_bytree = 1,
gamma = 0.5)
x 1:7])
y "Yes", 1, 0)
train.mat
这些准备工作完成之后,即可创建模型:
set.seed(1)
xgb.fit 75)
xgb.fit
## ##### xgb.Booster
## raw: 29 Kb
## call:
## xgb.train(params = param, data = train.mat, nrounds = 75)
## params (as set within xgb.train):
## objective = "binary:logistic", booster = "gbtree", eval_metric = "error", eta = "0.1", max_depth = "2", subsample = "0.5", colsample_bytree = "1", gamma = "0.5", silent = "1"
## xgb.attributes:
## niter
## callbacks:
## cb.print.evaluation(period = print_every_n)
## # of features: 7
## niter: 75
## nfeatures : 7
pred # summary(pred)# head(pred)# head(y)
在测试集上查看模型效果之前,先检查变量重要性,并绘制统计图。你可以检查3个项目:gain、cover和frequecy。gain是这个特征对其所在分支的正确率做出的改善,cover是与这个特征相关的全体观测的相对数量,frequency是这个特征在所有树中出现的次数百分比。以下代码会生成我们需要的输出:
impMatrix 2]], model = xgb.fit)
impMatrix
## Feature Gain Cover Frequency
## 1: glu 0.40000545 0.31701685 0.24509804
## 2: age 0.16177613 0.15685051 0.17156863
## 3: bmi 0.12074047 0.14691329 0.14705882
## 4: ped 0.11717240 0.15400327 0.16666667
## 5: npreg 0.07642331 0.05920866 0.06862745
## 6: skin 0.06389970 0.08682107 0.10294118
## 7: bp 0.05998254 0.07918635 0.09803922
xgb.plot.importance(impMatrix, main = "Gain by Feature")
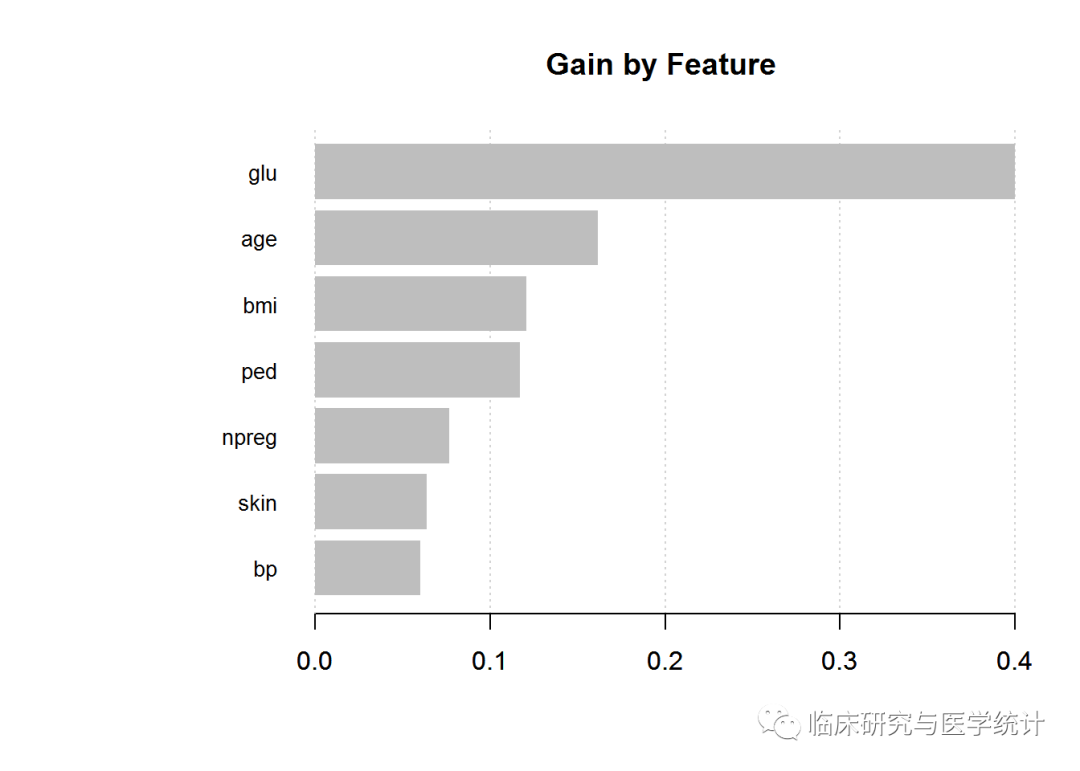
与其他方法相比,这个特征重要性图有什么不同吗?