我们将简要描述差异表达分析的主要步骤,这是RNA-seq数据分析中最为常规的任务。示例工作流程图中假设参考基因组是可用的。分析每一步,我们都会描述分析目的,一些典型的选项,输入和输出的文件,并指出可以找到详细步骤的完整章节。我们希望提供整个RNA-seq数据分析流程的概述,以便使用者可以看到各个步骤间是如何相互关联的。
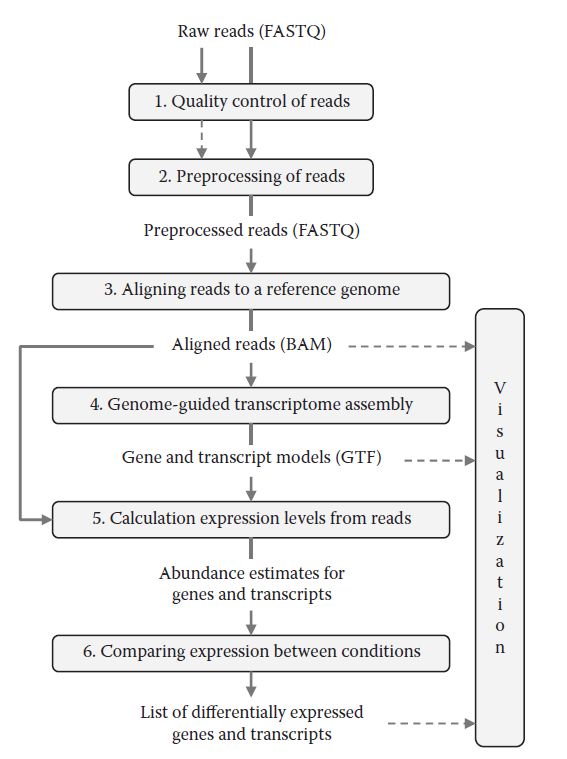
数据分析开始于原始的测序reads,通常是FASTQ格式,但有时也可以使用其他格式。当程序不支持其他格式时,reads必须重新转换为FASTQ格式。分析的第一步,进行普通的的质量控制分析。用于查看几百万个reads的整体质量。对reads进行扫描可以获得低置信度碱基,偏向性的核苷酸,测序接头及是否过度扩增等信息。此步骤的输出结果是基本统计数据,如reads数量和质量信息,这些数据将指导下一步中的预处理操作。
数据预处理的目的是从单个reads中移除低质量的碱基和人为的插入片段,如测序接头或文库构建所需的序列。建库实验所需的序列也可以被删除。例如,poly A尾巴可以被删除,它们会干扰随后的分析步骤。人为插入片段的另一个来源是存在于许多生物体中的微生物群体。从人类RNA组织样品中去除大肠杆菌基因组序列可能有助于随后的下游分析。一些reads也可能因为它们的长度而被去除。例如,成熟的miRNA序列长度为21-22nt,而测序reads长度为150nt。预处理之后,可以进行下一个数据分析步骤。
此步的目的是找到每个测序read在参考基因组上的对应点。如果还没有可用的参考基因组,reads可以被到匹配到一个转录组上。当reads匹配到参考基因组时,会产生一个序列比对文件。除了预处理的reads之外,在该步骤中还需要有参考基因组序列作为输入文件之一。序列拼接计算量是非常巨大的,因为有数以百万计的reads进行比对,基因组也非常庞大,拼接的reads必须非连续地进行匹配上去。因此,基因组序列常常被转换并压缩成索引文件以加快拼接。最常见使用的一个软件是Burrows-Wheeler转换工具。此步骤的输出是一个序列比对文件,它列出了匹配上的reads及其在参考基因组中的匹配位置。除了下游分析之外,也可以使用基因组浏览工具在基因组水平上进行可视化匹配上的reads。
一旦reads被拼接到基因组上,这个拼接文件就可用于发现未知基因和可变
剪接。基因相对于测序reads来说是非常大的。例如,哺乳动物成熟mRNA通常是1.5kb,而RNA-seq的reads则是100-250nt。因此,通常情况下不可能根据单个测序reads得到一个转录本的确切结构(转录起始位点,外显子-内含子区域,polyA位点)。更长的片段读取的测序平台,如PacBio系统的确可以读取一个完整的转录本,但目前读长较短的测序数据分析仍然占据主导地位。大多数外显子<200bp,因此需要通过匹配到基因组上的情况来重建可变外显子的使用和顺序,然后把短的片段之间连接起来。虽然表面上听起来比较简单,但是对计算有很高的要求,需要一些技巧性的解决方法。这一步输出的结果是基因和转录组模型。来自不同样品所组装的转录本通过参考注释被合并和组合,从而产生更完整的基因模型,然后被用于下一步的表达量分析。
通过数据分析产生的关键数据表格包含每个基因和转录本的reads数目。在此步骤中,一个单独的reads与某个基因是否相关联取决于匹配到基因组上的位置。未知基因和转录本的表达水平可以使用前一步获得的基因组模型进行定量。当处理具有详细注释文件的生物体如人类时,可以使用参考注释,从而将定量仅限于已知的基因和转录本。基因表达丰度估值可以用原始reads或以均一化单位(例如RPKM(每百万个reads中每个转录本的千个核苷酸所包含的reads数目)或FPKM(每百万个匹配上的reads中每千个核苷酸所包含的测序片段数目)进行呈现。测序Reads匹配到基因上的数目以及在不同特征类型的基因组上的信息,也可以作为重要的质量控制参考标准。在这一步,数据变成简单的基因列表及其reads数目或RPKM / FPKM值。










