新媒体管家
微生物与人类生活息息相关,除了在环境中遇到各类微生物之外,我们的身体也离不开微生物。据统计,成人肠道含有100万亿微生物,其基因数是人基因数的100倍,总重量在1.5 kg左右(Jesse D, 2013)。
因此,K Ray曾诙谐地表明,“我们更像是微生物而不是人,我们与肠道菌群之间并非朋友关系而是更亲近—我们已经‘嫁给’它们”(K Ray, 2012)。
图1 我们更像是嫁给微生物
(图片来源《
Nature Reviews Gastroenterology & Hepatology
》)
既然“嫁给”了微生物,我们就要对微生物有充分的了解,宏基因组技术则是了解微生物群落结构和功能的利器。
随着基因组测序成本的降低,更多的专家学者倾向于通过大数据来分析物种多样性、功能基因、构建代谢通路等。然而现在,利用宏基因组大数据量却可以完成传统技术无法解决的问题,即Contig Binning的分析方法解析那些无法纯培养微生物的基因组。
Contig Binning是基于宏基因组测序序列,将组成相似或丰度一致的Contigs聚类到同一物种从而完成单菌的草图组装,再利用常规的生物信息分析的方法解析基因组的功能。
因此,对于那些难以培养的微生物,利用Contig Binning的生物分析方法可获得它们的基因组草图。
以下是近几年发表的用Contig Binning的分析方法组装难培养微生物的文章,可供大家参考:
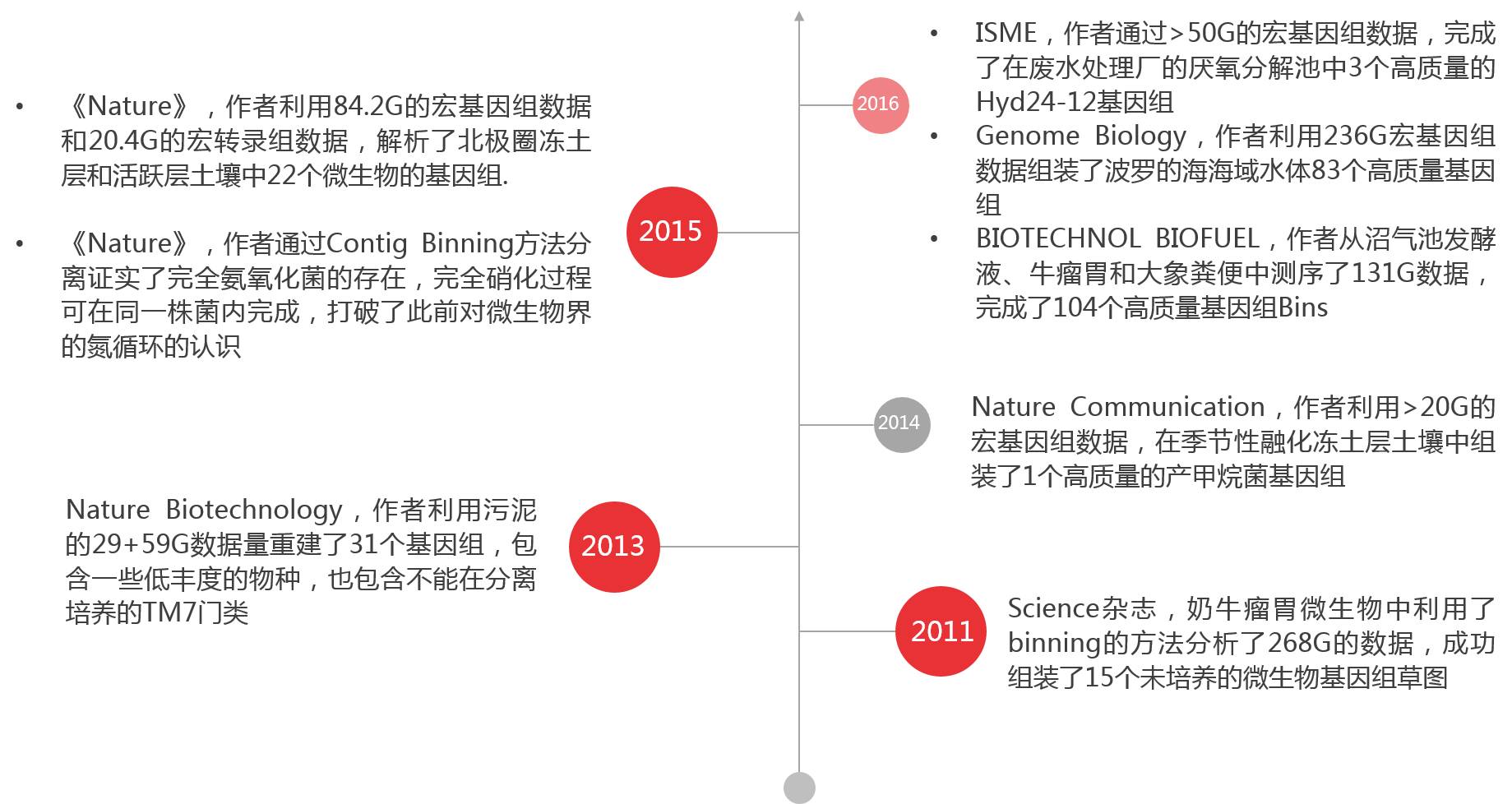
图2 基于
Contig Binning分析方法的系列文章
既然Contig Binning的分析策略可以完成难培养的微生物基因组草图,那它的研究思路究竟如何?
事实上,无论是环境微生物还是人体微生物,都可以通过Contig Binning的方法进行研究,并得到高质量的微生物基因组。
早在2012年就有专家学者针对肠道微生态样本进行了Contig Binning组装,并获得了高质量的微生物基因组。
Mads曾在《Nature Biotechnology》发表了一篇题为《Genome sequences of rare, uncultured bacteria obtained by differentialcoverage binning of multiple metagenomes》的文章,并呈现了Contig Binning的组装思路:
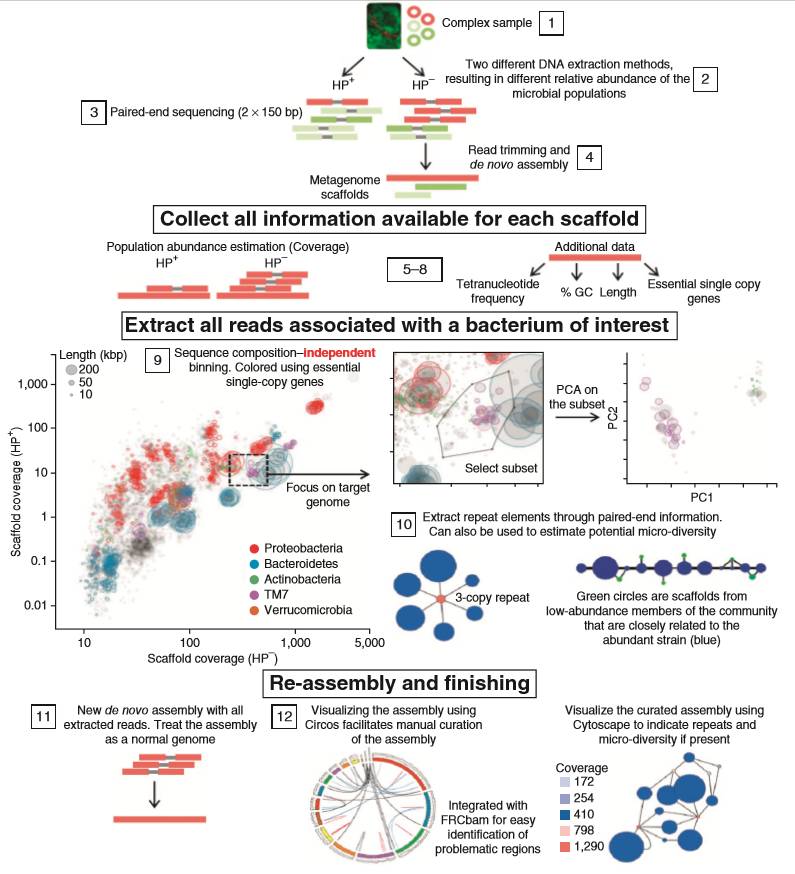
图3 Contig Binning的组装思路
(图片来源《
Nature Biotechnology
》)
Step1:
样品准备
Step2:
两种不同的方法提取DNA(热酚提取与非热酚提取),因此不同的微生物组分会有不同的相对丰度
Step3:
PE 150测序(HP+和HP-分别获得29G和57G高质量测序数据)
Step4:
宏基因组组装,得到非冗余的scaffolds序列
Step5-8:
计算两种提取方法中每个scaffolds的丰度,并进行均一化处理;同时计算Teltranucleotide频率、GC含量、是否是保守的单拷贝标记基因(如16S rDNA序列)作为序列聚类的参考标准
Step9:
以两种提取方法获得的scaffolds丰度构建坐标系,依据scaffolds的丰度进行Contig Binning聚类,Contig Binning的集合就代表了假定的某一微生物群落的基因组,其中圆圈的大小代表序列的长度,不同颜色代表单拷贝基因。由于不同的物种可能会有同一丰度的基因集合,因此Contig Binning的聚类结果还需进行Teltranucleotide频率的主成分分析,以区分不同物种
Step10:
从测序数据中筛选重复元件或多拷贝基因
Step11-12:
筛选所有的与Contig Binning聚类集合相关的reads,重新进行基因组de novo组装,获得一个标准的基因组草图。可根据保守的单拷贝基因对组装结果进行验证,明确组装或其他的基因组结构问题,并通过Cytoscape对重复序列和微生物多样性代表序列进行可视化
作者通过Contig Binning的分析方法,最终获得了31个细菌基因组,包括相对丰度在1%以下的物种基因组,并对31个细菌物种进行了门水平的分类。
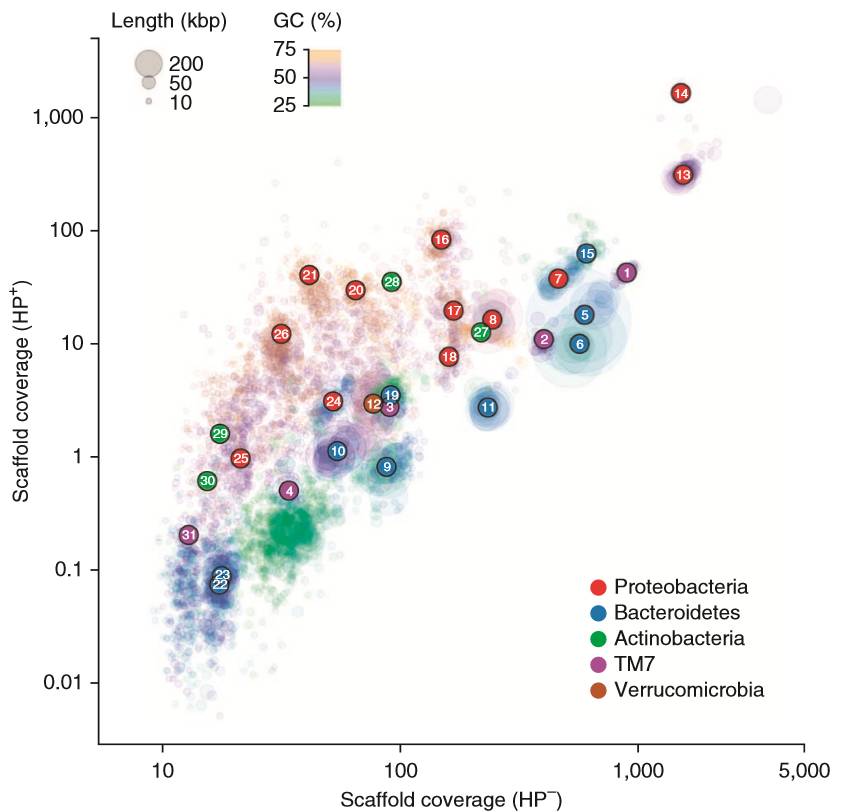
图4 通过Contig Binning获得的31个细菌基因组
(
图片来源《Nature Biotechnology》
)
同样,Mohamed利用宏基因组数据,研究了红海海域8个位置的微生物群落,通过Contig Binning的序列聚类和分析,获得了136个微生物基因组(Mohamed
et al.
, 2016)。与Mads稍有不同的是,Mohamed主要将Teltranucleotide和序列相对丰度作为Contig Binning的聚类的参考指标,具体的分析策略如下:
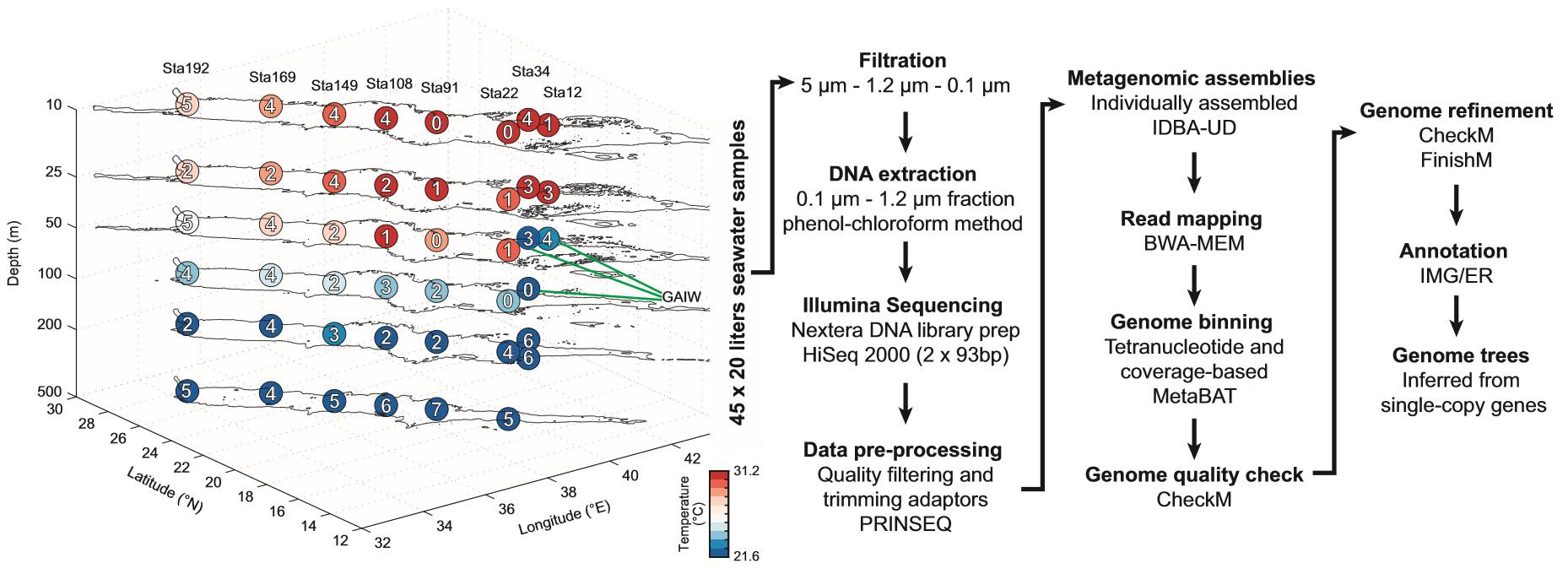
图5 Mohamed在红海海域的取样位置及Contig Binning的分析策略
(
图片来源《Scientific Data》
)
对组装获得的136个微生物基因组,Mohamed分别进行了古菌和细菌的分类分析。对于古菌而言,作者依据122个单拷贝标记基因构建物种进化树;对于细菌而言,作者依据120个单拷贝标记基因进行物种分类分析,结果如下:
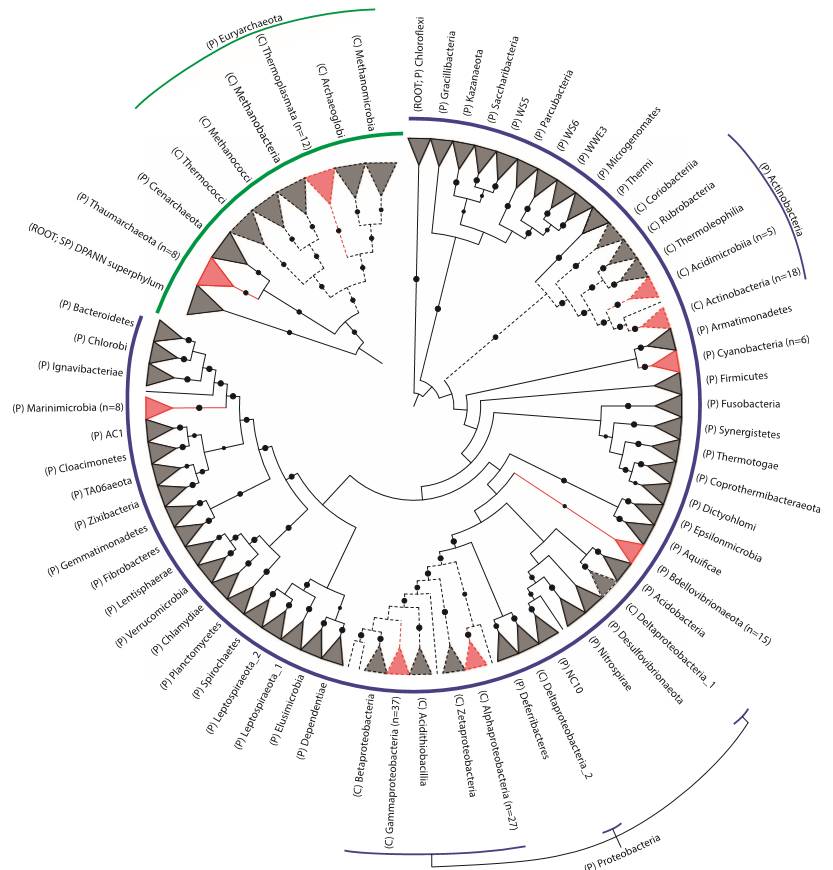
图6 Mohamed对组装的古菌和细菌构建物种进化树
(
图片来源 《Scientific Data》
)
由于Contig Binning的组装策略可以帮助科研工作者探索样品中难以培养的微生物群落,因此自从Contig Binning的组装策略问世以来,很多专家学者都陆陆续续对Contig Binning的组装方法展开了研究。
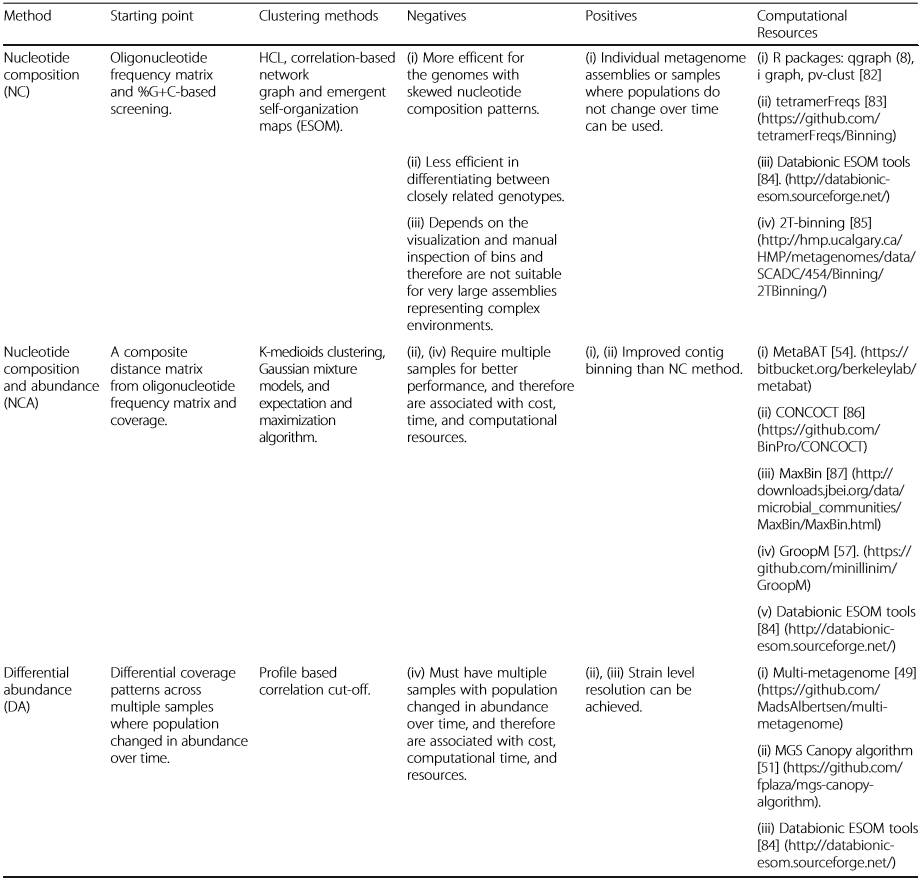
Microbiome期刊于2016年发表了一篇综述,认为Contig Binning的组装策略主要分为3类:
核酸组成(NC)
、
核酸组成与丰度(NCA)
、
积分丰度(DA)
等,然而各个方法都有各自的优缺点(Naseer Sangwan, 2016)。将核酸的组成与序列的丰度相结合被认为是一种比较好的策略,既能保证Contigs Binning的效果,也能相对节约计算资源。具体信息如下表所示:
表1 Contig Binning的组装策略
(图表来源《Microbiome》)
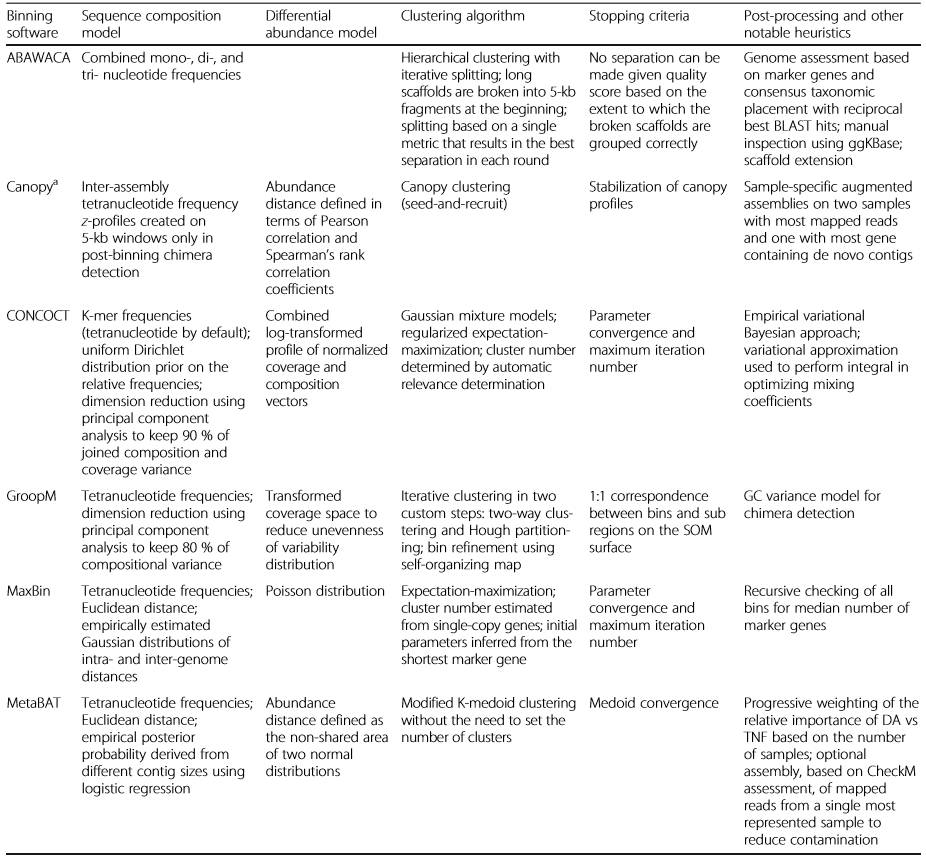
另外,这篇综述还汇总了用于Contig Binning组装的软件及其功能介绍,
其中MetaBAT就是Mohamed在《A catalogue of 136 microbial draftgenomes from Red Sea metagenomes》中使用的Contig Binning组装软件:
表2 Contig Binning的组装软件
(
图表来源《Microbiome》
)
同样的,该篇综述对Contig Binning的整体思路做了总结。细细品味,该篇综述所总结的思路其实与Mads在《Genome sequences of rare, unculturedbacteria obtained by differential coverage binning of multiple metagenomes》文章中所提倡的思路有不谋而合之处。
两种思路都提倡依据序列丰度和核苷酸组分分析(核苷酸频率、GC含量、必要的单拷贝基因)来进行Contig Binning的基因组组装。具体思路可参考下图:
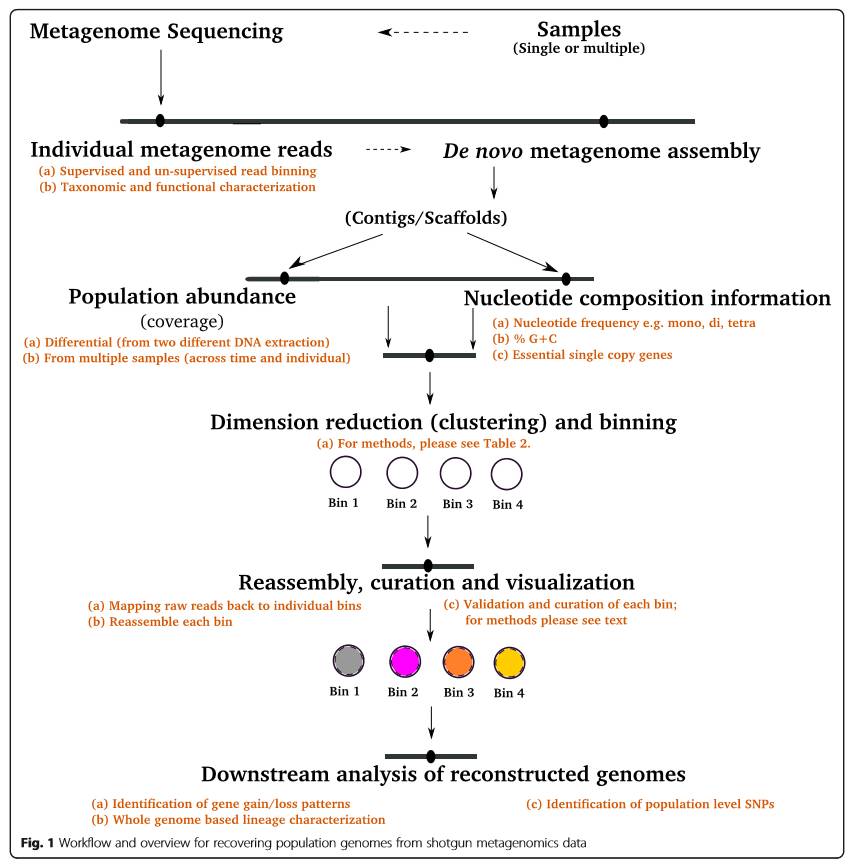
图7 Contig Binning的整体思路
(
图片来源 《Microbiome》
)
需要值得注意的是,对于不同的研究,或者依据不同的样本量的大小,或者对于不同的样本类型,选择Contig Binning的组装策略都会有细微的差异。因此,我们在选择Contig Binning来组装无法培养的微生物基因组时也需谨慎。
参考文献:
[1] Jesse D. Aitken and Andrew T. Gewirt. Toward understanding and manipulating the microbiome to treat intestinal disease. 2013
[2] K Ray .Gut microbiota: married to our gut microbiota. Nature Reviews Gastroenterology & Hepatology. 2012
[3] Mads Albertsen et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nature Biotechnology, 2012
[4] Mohamed et al. A catalogue of 136 microbial draft genomes from Red Sea metagenomes. Scientific Data, 2016
[5] Naseer Sangwan. Recovering complete and draft population genomes from metagenome datasets. Microbiome, 2016
关于金唯智高通量测序服务
金唯智配备有完整的一代、二代和三代测序平台,可为客户提供多种测序读长、通量和时间周期选择,满足生命科学研究用户对成本和速度的差异化需求。通过自主研发和合作开发,在全长转录组、全长16S分析及基因组学分析等方面形成了独特的技术优势。凭借持续的研发创新,全球化的运营管理和先进的设备平台,为科研人员在基因组学研究、精准医疗与其它应用领域的研究提供最全面的基因组学解决方案。
金唯智拥有全球领先的高性能计算平台以及数据处理能力,采用EMCIsilon集群存储系统,拥有2PB的存储容量。携手阿里云、微软云,协同打造大数据分析的云计算能力,可快速、高效地读取高质量的测序数据,并完成精准、稳定的数据分析及交付,大幅度提高生命科学研究的速度和效率。
我是魔法师GENE
金唯智的形象大使
秉持财富唯有智慧创造
为您传递基因组学服务咨询
助力您在生命科学领域的研究





