正文
psRobot:植物小RNA分析系统

简介
官网:http://omicslab.genetics.ac.cn/psRobot/
PsRobot是中科院遗传发育所王秀杰组的作品,主要实现小RNA的mapping,miRNAs前体和成熟体的预测、降解组分析等功能。发布不到5年,截止17年7月25日,Google scholar统计引用91次(不乏Nature, Sicence文章),psRobot网站统计访问量498139,独立IP数13603,执行分析任务321001次。
注:论文的一作吴华君博士目前在哈佛做博后,完成本软件的本地版编程;共同一作马英克博士负责本软件的webserver开发及维护(兼职于易汉博,最新发表博文NGS基础 - 参考基因组和基因注释文件);三作正是生信宝典创始人陈同博士,负责本软件的本地版部署;当年我参与了软件的测试工作,文末致谢部分有我的名字。
功能概述
本程序主要分为在线版和本地版,但功能不完全相同,即适合生物学家在线分析,又适合生信专家本地大规模分析。
在线版主要功能
小RNAs前体预测:基于测序的small RNA和参考基因组,预测新的miRNAs; http://omicslab.genetics.ac.cn/psRobot/stemloop_1.php
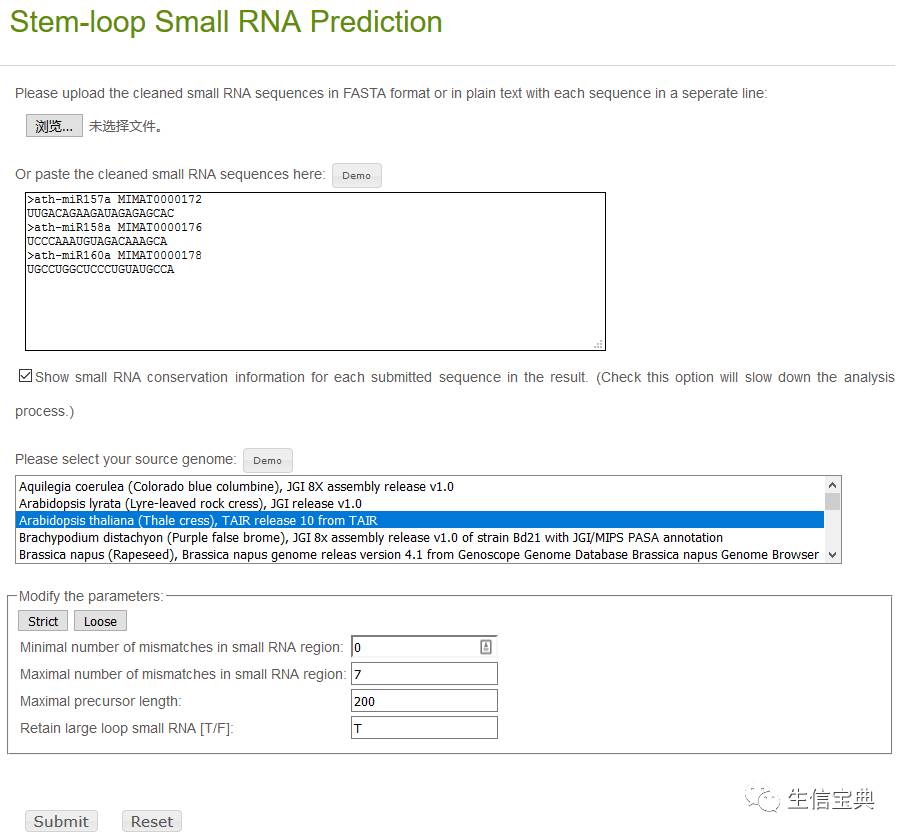
小RNAs靶位点预测:基于新发现或已经发表的小RNAs,预测指定物种转录本中的靶位点。 http://omicslab.genetics.ac.cn/psRobot/target_prediction_1.php
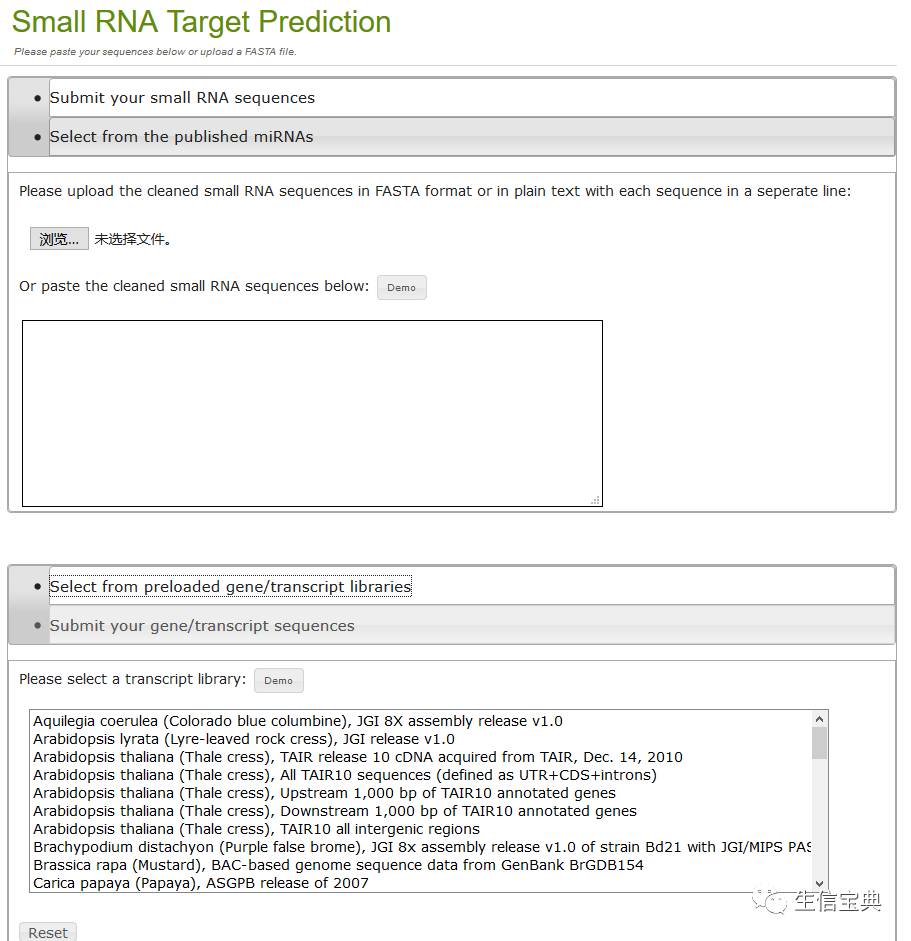
网页操作方法极简便,只需上传或输入需要分析的小RNAs或mRNA序列,选择对应物种即可,使用方法不再赘述。第一次使用点网页中的Demo看测试数据格式及输出结果,快速上手。
本地版主要功能
本地版主要分为四个功能模块:
psRobot_map:将miRNAs mapping到参考基因组;采用C语言编写,虽然不支持错配,但对于small RNA应该足够了。
psRobot_mir:基于测序的small RNA和参考基因组,预测新的miRNAs;
psRobot_tar:预测小RNAs在转录本中的靶位点。
psRobot_deg:降解组分析,对预测靶位点的进一步实验验证。
近几年发表的小麦的Nature,Sciences文章中small RNA分析部分均采用此软件。而且此软件,也被很多生信大公司采用作为标准分析方法,如华大基因、诺禾致源、易汉博等。
使用方法
最新帮助文档下载 http://omicslab.genetics.ac.cn/psRobot/program/WebServer/psRobot_manual_v1.2.pdf
本文以介绍本地版的安装和使用为主,软件我这个月还用过,安装使用请以本文为准,新于官方文档。
本文主要详解本地版的使用实例,并且以比较复杂的大麦为例,采用前3个模块进行序列比对、预测新的miRNAs和靶位点的预测。
安装在Ubuntu 16.04上
# 依赖软件mfold3.6安装
wget http://unafold.rna.albany.edu/download/mfold-3.6.tar.gz
tar xvzf mfold-3.6.tar.gz
cd mfold-3.6/
./configure
make
sudo make install
nafold # it work
# PsRobot软件 安装
wget http://omicslab.genetics.ac.cn/psRobot/program/WebServer/psRobot_v1.2.tar.gz
tar xvzf psRobot_v1.2.tar.gz
cd ../psRobot_v1.2
sudo ./configure
make
sudo make install
source /mnt/bai/public/.bashrc
安装问题参考 Linux学习 - 命令运行监测和软件安装 Linux学习-环境变量和可执行属性
原始数据格式转换和mapping
# 干净的sRNA fastq文件转换为软件要求格式
zless seq/sample.fq.gz | grep '^[AGCT]' | sort --parallel=8 | uniq -c | sed 's/ *//' | awk '{print 1}' > seq/sample.sRNA
# 多样品合并、按RPM丰度和长度选择,需要此步脚本分享教程至朋友圈并联系微信yongxinliu索要
sRNA_merge.pl -i 'seq/*.sRNA' -o temp/merge.sRNA -r 1 -s 18 -l 26
# 转换sRNA序列为fasta格式
awk '{print ">"NR"_"$$1}' temp/merge.sRNA > temp/merge.fa
# fasta格式转换为psRobot要求格式
awk '{print NR"_"1}' temp/merge.sRNA > temp/merge.psmap # format sRNA to psmap
# 比对序列至参考基因组
psRobot_map temp/merge.psmap barley.fa temp/merge.psmaping
新miRNAs的预测
# 一定要新建目录,会生成很多文件
mkdir -p psRobot
cd psRobot
# 基于所有样品sRNA和基因组预测新miRNA,运行时间长,建议后台运行
nohup psRobot_mir -s ../temp/merge.sRNA -g ../barley.fa &bg
结果会有3个文件,如下:
Final_PreDict_miRNA_samp.StarInfo
包括预测的miRNAs基本信息
# 1ID_LociNumber_LociOrder_Length_Count 2miRNAs 3No.miRNA.cluster 4miRNA* 5miRNA*Seq 6PrecursorLocation 7Precursor
Sr15842_1_1_21_419 TAAGATTTGTAGGTGATTGGG 1 - - bgh_dh14_v3.0_supercontig_005464:163257:163389:+ gatttttcgatTAAGATTTGTAGGTGATTGGGtgtatgcttgcgttatgtctctaagccagagtgaatttccataaatttcaaaaagtgtgagggctagagcaacaattagtcgctgcgaagctcgttgtatt
Sr47712_3_1_22_3075394 TGAAGCTGCCAGCATGATCTGA 2 Sr47287_2_1_21_1485 aggtcatgtggcagcttcatt chr4H:590940935:590941057:- ccgcaagtagaTGAAGCTGCCAGCATGATCTGAaagctatgctgcatgtcgatctcgatggtcgtctccatccagattcaagagcatggccggcaatcaggtcatgtggcagcttcattttct
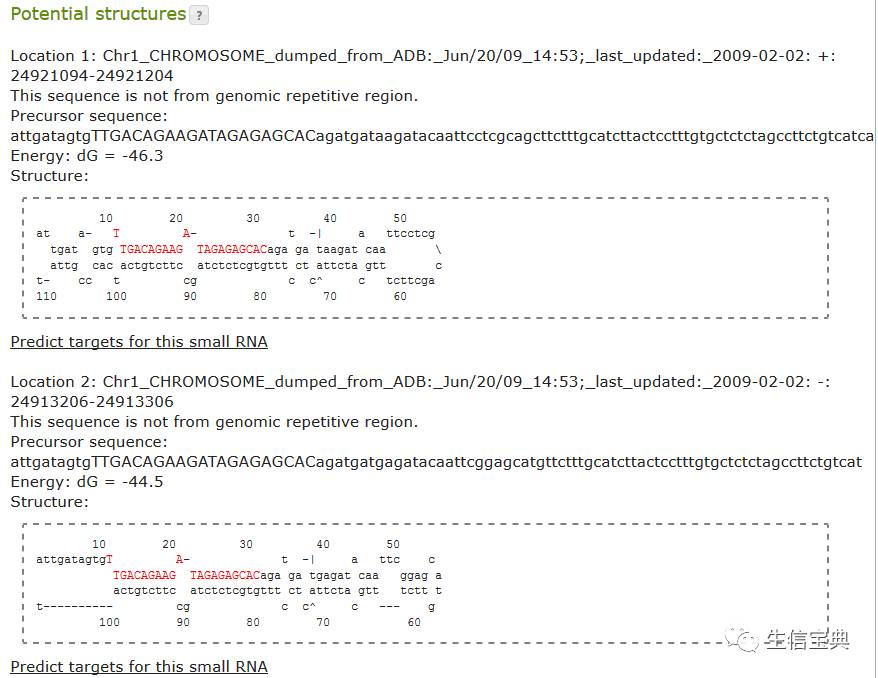
Final_PreDict_miRNA.Struc
包括预测的二级结构信息,如Sr20904_5_3_22_1342||91||10为样品ID,后位有数字为其上或下游碱基的数量,即其在前体中的位置;-24.10为折叠自由能;下面的为二级结构图。
Sequence 1 Structure 1
Folding bases 1 to 123 of Sr20904_5_3_22_1342||91||10
dG = -24.10
10 20 30 40 50 60
aaattggattaagaagatt t c - a - -| t tta ac cattaa
gtc gttgc atttct cgt tgt c tcc gct act ct \
cgg TAACG TAAGGA GCG ACA g agg tga tga ga a
aaaggtt------------ T A T G T a^ c cc- aa cctaac
120 110 100 90 80 70
3. Final_PreDict_miRNA.Reads
有每个miRNAs区详细的mapping结果。
>Sr30070_6_1_19_4858 chr1H|13342638|13342720|+
cggcagctgcgCTCGGCGGGGCAGCGTGCAgagggacttcgtccggcgcgctcctccgtcgagcgtggctccggtgacgcgtt 8820
***********CTCGGCGGGGCAGCGTGCA***************************************************** 6 19 4858
-----------CTCGGCGGGGCAGCGTGCA----------------------------------------------------- 6 19 4858
-----------CTCGGCGGGGCAGCGTGCAG---------------------------------------------------- 6 20 577
-----------CTCGGCGGGGCAGCGTGCAGA--------------------------------------------------- 6 21 600
------------TCGGCGGGGCAGCGTGCA----------------------------------------------------- 6 18 1450
------------TCGGCGGGGCAGCGTGCAGAG-------------------------------------------------- 6 21 1335
miRNAs靶基因的预测
此步需要fasta格式的miRNAs序列,可以是miRBase上发表已知的,也可以是上面预测的。本文以上面预测的miRNAs为例,靶基因库为物种的cDNA序列。
cd ..
# 筛选新miRNAs
cut -f 2 Final_PreDict_miRNA_samp.StarInfo|sort|uniq| awk '{print ">"NR"\n"$0'} > predict_miRNA.fa
# 进行靶位点预测
nohup psRobot_tar -s predict_miRNA.fa -t temp/barely_cdna.fa -o temp/merge_miRNA_barely.gTP -p 20 &bg
输入miRNAs和cDNA,输出gTP文件可less查看,支持多线程,运行时间长,建议后台运行。结果示例如下:
>6429_311 Score: 2.5 HORVU1Hr1G055350.18 cdna chromosome:Hv_IBSC_PGSB_v2:chr1H:405769196:405773487:-1 gene:HORVU1Hr1G055350 gene_biotype:protein_coding transcript_biotype:protein_coding
Query: 1 ACTAATGACGCATTTGTAGATGGT 24
||||||||||||::|:||**||||
Sbjct: 3095 TGATTACTGCGTGGATATAGACCA 3072
降解组数据分析
最近没做这方面的分析,需要使用的看帮助文档就行了,写的已经非常详细了。
Reference
[Wu HJ, Ma YK, Chen T, Wang M, Wang XJ. (2012) PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. DOI:10.1093/nar/gks554.] (http://nar.oxfordjournals.org/content/40/W1/W22)
这么好的在线数据库,我也想要
如果实验室有了不少测序数据
或
研究用到了不少公共数据
或
开发了本地软件
想整合起来
共享使用
快速查询
可视化
在线分析
抑或是
发表一篇文章
最大限度发挥数据的价值
怎么办?
易汉博团队
不断迭代更新
已有比PsRobot更好的设计
更适合高通量数据的功能
操作简单
全局搜索
模糊匹配
通路查询
互作网络
交互式可视化
基因组浏览器
。
。
。

加入易汉博,人生不白活

关注生信宝典,换个角度学生信

关注宏基因组,再专业一点










