中文翻译:栗增博士
-FDA批准Zykadia用于ALK阳性的一线转移性非小细胞肺癌的扩大治疗
-Mundipharma公司的药物Mundesine在日本上市
-Alunbrig在美国开始接受订单
-FDA批准了 Kisqali与Femara的组合制剂
-Rydapt在美国开始销售
-FDA批准Rydapt 用于治疗新确诊的带有FLT3突变的急性髓系白血病
-FDA加速批准了Alunbrig用于治疗ALK阳性突变的非小细胞肺癌
-FDA加速批准了Keytruda的新适应症并会对后续适应症申请进行优先评审
-Imfinzi在美国上市销售
-FDA批准Keytruda用于局部晚期或转移性尿路上皮癌特定患者的治疗
-欧盟批准dinutuximab beta用于治疗高危神经母细胞瘤
-FDA批准Bavencio用于治疗晚期尿路上皮癌
-FDA加速批准了Imfinzi用于治疗尿路上皮癌
-
Actemra成为FDA批准的治疗巨细胞动脉炎的首个药物
-
FDA批准Tymlos用于绝经后女性患者的骨质疏松症的治疗
2017年5月29日,FDA批准Zykadia用于ALK阳性的一线转移性非小细胞肺癌的扩大治疗
美国FDA批准扩大使用Zykadia(Ceritinib)用于治疗一线转移性非小细胞肺癌(NSCLC),适用病人为通过FDA批准的检测方法确认的ALK阳性患者。Zykadia于2014年首次获得加速批准用于疾病进展或对crizotinib不耐受的ALK阳性的转移性NSCLC的患者治疗(参见汤森路透药物快讯2014年4月30日)。2017年1月,FDA授予了Zykadia突破性疗法认定,用于一线治疗ALK阳性的脑部转移性NSCLC患者,并向ALK阳性转移性NSCLC一线治疗颁发了优先审评资格(参见汤森路透药物快讯2017年2月23日)。Zykadia的一线疗法获批是基于一项名为ASCEND-4的全球范围的开放标签的随机多中心III期临床试验的结果(ClinicalTrials.gov编号NCT01828099)。研究表明,Zykadia组患者的中位无进展生存期(PFS)为16.6个月,显著优于使用标准培美曲塞-铂化疗方案后维持治疗患者的8.1个月。Zykadia组脑转移患者的总体颅内缓解率为57%,优于化疗患者的22%。Zykadia组的全身总体缓解率为73%。FDA同时批准了用于检测ALK基因重组的方法来帮助判断患者最合适的治疗方案。(诺华公司新闻稿)
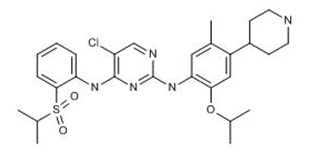
图为Ceritinib结构
2017年5月25日,Mundipharma公司的药物Mundesine在日本上市
Mundipharma公司宣布旗下药物Mundesine(forodesine hydrochloride,100mg胶囊)已经在日本上市。Mundesine是一个嘌呤-核苷磷酸化酶抑制剂,日本于2017年3月首次批准其对复发性或难治性外周T细胞淋巴瘤的治疗。自上世纪90年代初的标准一线疗法以来,非霍奇金淋巴瘤患者的治疗选择尚未取得进展。Mundesine作为单一的口服疗法预计将减少患者的负担。Forodesine hydrochloride是由BioCryst Pharmaceuticals开发的小分子化合物,在2008年被认定为孤儿药。(Mundipharma公司新闻稿)。
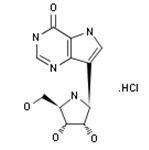
图为Forodesine hydrochloride结构。
2017年5月15日,Alunbrig在美国开始接受订单
McKesson Specialty Health公司宣布Alunbrig(brigatinib)日前开始由旗下药房在美国销售。Alunbrig是一个近期刚刚被FDA批准的,用于治疗疾病进展或对crizotinib不耐受患者的ALK阳性的转移性非小细胞肺癌的小分子口服药(参见汤森路透药物快讯2017年5月2日)。Brigatinib由武田制药的全资子公司Ariad Pharmaceuticals生产(McKesson Specialty Health公司新闻稿)。

图为Brigatinib结构
2017年5月10日,FDA批准了 Kisqali与Femara的组合制剂
FDA宣布批准了诺华制药的Kisqali-Femara组合制剂(ribociclib片剂+letrozole片剂)用于绝经后女性激素受体阳性/人类表皮生长因子受体2阴性(HR+/HER2-)晚期或转移性乳腺癌的治疗。依据这一批准,医生今后可以灵活的开具含Kisqali的组合处方或Kisqali与任意芳香酶抑制剂的搭配处方。此次获批的组合制剂有3种规格:分别为ribociclib 600 mg + letrozole 2.5 mg;ribociclib 400 mg + letrozole 2.5 mg和ribociclib 200 mg + letrozole 2.5 mg,并将于本月晚些时候上市销售。Kisqali在今年3月被批准和芳香酶抑制剂联合用药用于治疗(HR+/HER2-)晚期或转移性乳腺癌(参见汤森路透药物快讯2017年3月14日)。Femara是一种已被批准的用于绝经后妇女HR+或未知的晚期乳腺癌治疗的芳香酶抑制剂。(诺华公司新闻稿)。
上图为Letrozole结构
上图为Ribociclib结构
2017年5月4日,Rydapt在美国开始销售
McKesson Specialty Health公司的抗肿瘤药品服务公司Biologics宣布诺华公司的药物Rydapt(midostaurin)日前开始在美国国内接受订购。Rydapt近日刚被FDA批准用于新确诊的急性髓系白血病(AML)成年患者的治疗,这些患者需要利用FDA批准的检测方法确认FLT3突变阳性,同时联合标准的阿糖胞苷和道诺霉素疗法和阿糖胞苷巩固疗法(参见汤森路透药物快讯2017年5月2日)。Rydapt还被批准用于治疗侵袭性全身性肥大细胞症,系统性肥大细胞症与血液学肿瘤或肥大细胞白血病(McKesson Specialty Health公司新闻稿)。
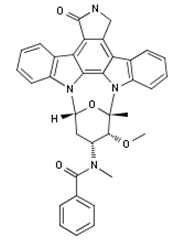
图为Midostaurin
2017年5月2日,FDA批准Rydapt 用于治疗新确诊的带有FLT3突变的急性髓系白血病
FDA批准了诺华制药的Rydapt (midostaurin)用于新确诊的带有FLT3突变的急性髓系白血病,并与化学疗法联合用药,即联合标准的阿糖胞苷和道诺霉素疗法和阿糖胞苷巩固疗法。FDA同时批准了Invivoscribe Technologies公司的伴随诊断试剂盒LeukoStrat CDx FLT3用于测定患者的FLT3突变。该药的疗效与安全性评价数据来源于一项有717名新确诊患者参与的随机临床试验(ClinicalTrials.gov编号NCT00651261)。在该研究中,接受midostaurin与化疗联合用药的患者的生存期优于仅接受化疗的患者,尽管其平均存活率目前还不能准确统计。同时,接受联合用药的患者由于没有出现特定的并发症而参与该研究的时间更长(中位数8.2个月),单纯化疗的患者由于在60天内出现疾病恶化或死亡,参与该研究的中位时间仅为3个月。Midostaurin的常见副作用包括:发热性中性粒细胞减少,恶心,粘膜炎,呕吐,头痛,瘀伤,肌肉骨骼疼痛,出血,与医疗器械相关的感染,高血糖和上呼吸道感染。Rydapt也被批准用于成人罕见血液疾病如侵袭性全身性肥大细胞症,系统性肥大细胞症与血液学肿瘤或肥大细胞白血病。此次获批是依据两项开放标签的单臂多中心临床试验(ClinicalTrials.gov编号NCT00782067和NCT00233454)。Rydapt的疗效是建立在确认完全缓解的基础上,加上六个疗程标准治疗的不完全缓解(89名患者)。分析表明整体响应率为21%。其疗效也经由2013 IWG-MRT-ECNM共识标准进行了事后分析(115名患者),其完全或部分缓解率为17%。midostaurin最常见的不良反应为恶心、呕吐、腹泻、水肿、肌肉骨骼疼痛、腹痛、乏力、上呼吸道感染、便秘、发热、头痛、呼吸困难。FDA 授予了该上市申请优先审评资格、快速通道审评资格(用于肥大细胞增多症)及突破性疗法资格(用于急性髓系白血病)。(FDA新闻稿,诺华公司新闻稿)。
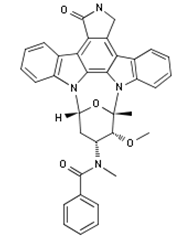
图为Midostaurin结构
2017年5月2日,FDA加速批准了Alunbrig用于治疗ALK阳性突变的非小细胞肺癌
武田制药的子公司Ariad Pharmaceuticals的新药Alunbrig(brigatinib)获得了FDA的批准,用于治疗疾病进展或对crizotinib不耐受患者的ALK阳性的转移性非小细胞肺癌。此次获批是基于一项非对照的开放标签的双臂临床II期ALTA试验(ClinicalTrials.gov编号NCT02094573)。用药组患者在接受推荐剂量每天90mg持续7天后再使用每天180mg的剂量,其中54%的患者获得了经调查认定的客观应答,中位答应时间为独立评审委员会评估的13.8个月和研究者评估的11.1个月。此外,在推荐剂量方案下的脑转移可测量的患者中,67%(18例)获得经证实的颅内客观应答。在23例发生颅内应答的患者中,78%来自90mg剂量组且其中的68%获得了持续4个月以上的持续缓解。主要的副作用(发生率超过25%)主要有:恶心,腹泻,疲劳,咳嗽和头痛。最常见的严重不良反应是肺炎和间质性肺病和肺炎。8例患者(3.7%)发生致死性不良反应,包括肺炎(2例)、猝死、呼吸困难、呼吸衰竭、肺栓塞、细菌性脑膜炎和尿路脓毒血症(各1例)。在欧洲,该药的上市申请已在今年2月提交至EMA(参见汤森路透药物快讯2017年2月7日)。(武田公司新闻稿)。
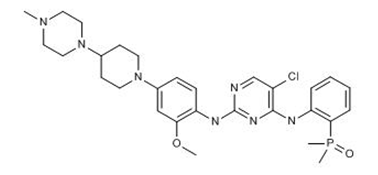
图为Brigatinib结构
2017年5月24日,FDA加速批准了Keytruda的新适应症并会对后续适应症申请进行优先评审
FDA加速批准了Merck 公司的Keytruda(pembrolizumab)对成人和儿科患者的不可切除或转移性实体瘤的治疗,其肿瘤中含有微卫星高不稳定性(MSI-H)或错配修复缺陷(dMMR)的生物标志物。这一指标涵盖了在先前治疗后取得进展的实体瘤患者,和没有令人满意的替代治疗方案的患者以及在接受过某些化疗药物如氟胞嘧啶、奥沙利铂和伊立替康治疗后病情有所改善结直肠癌的患者人群。这是FDA首次批准基于共同生物标志物的癌症治疗,而不受肿瘤起源身体哪个部位的限制。为了验证Keytruda治疗携带MSI-H或者dMMR实体瘤患者的有效性和安全性,研究团队开展了5项单臂临床试验,共招募了149名癌症患者,涉及15种肿瘤类型。其中,最常见的癌症是结直肠癌、子宫内膜癌和其它消化道癌症。Pembrolizumab的主要临床终点为总缓解率和持续应答时间。在全部149名患者中,39.6%发生了完全或部分的应答,其中的78%持续应答时间超过了6个月。在另一个报道中,FDA还接受了Keytruda的一项补充申请,用于已经接受了两线或更多的化疗的复发或晚期胃食管结腺癌的治疗,并授予了Keytruda在2017年9月22日到期的优先评审权。该项申请基于了一项名为KEYNOTE-059的临床II期试验的第一阶段结果,该项研究评估了pembrolizumab对已经接受两线或更多的化疗的复发或晚期胃食管结腺癌的重度预治疗患者的疗效(ClinicalTrials.gov编号NCT02335411)。该研究的数据将于下月对外公布。(Merck公司新闻稿;FDA新闻稿)。
2017年5月24日,Imfinzi在美国上市销售
阿斯利康公司的Imfinzi(durvalumab)已经在美国上市销售。McKesson Specialty Health公司作为被选定的经销商负责该药的分销。FDA在在早些时候批准了Imfinzi用于治疗晚期或转移性尿路上皮癌患者。这些患者在先前的铂类化疗中或化疗后病情依旧出现进展,或在接受辅助或新辅助铂类化疗的12个月内疾病出现进展(参见汤森路透药物快讯2017年5月2日)。(McKesson Specialty Health公司新闻稿)。










