
嗨嗨嗨!馆长我又来啦!家人们有什么困难放心大胆找馆长!这不最近有人反映做免疫组化实验好痛苦,不知道有没有不做实验也可以发高分的方法,馆长为此可是千辛万苦给大家带来这篇绝佳的高分文章!快来看看吧!
来自温州医科大学的江春辉团队,通过
单细胞RNA测序、差异性分析、GO富集分析、加权基因共表达网络分析、PPI网络、免疫浸润分析以及基因调控网络分析
等一系列方法,成功拿下5分+期刊!看到这么多方法肯定会有人说看的都头晕,别急!让馆长带大家梳理梳理~
1、脓毒症的转录因子+RNA测序!
在这篇文章中,作者先从
GEO数据库
进行数据采集,并通过
单细胞RNA测序,进一步从数据集中获取脓毒症样本,丰富详细的样本大大增加了文章结果的可靠性! 随后,为了得到各种条件下差异表达基因的交集,作者进行了
差异表达分析
。
(
PS:大家在生信分析过程中是否常常遇到电脑卡顿、无法出结果的问题?不用担心,强大的生信云服务器让您无后顾之忧!敬请扫码联系!
)
2、GO富集分析+WGCNA+PPI网络!
接下来,作者通过
GO分析
并结合
WGCNA
和
PPI
识别了其中关键的免疫相关中枢基因,并通过
ROC曲线分析
与
ELISA
验证了NLRC4在脓毒症中高表达的事实。环环相扣的研究方法并配上谨密的结果验证,馆长认为这也是这篇文章能得到审稿人信赖的一方面哦!
3、单细胞RNA测序与基因调控网络分析!
最后,作者还创新的使用
单细胞RNA测序技术
结合
基因调控网络分析(GRN)分析
进一步探究了脓毒症中转录因子活性和差异,完善了对脓毒症免疫应答机制的研究。
(
如果在生信选题、实验分析上有疑问,欢迎扫码与馆长联系!馆长为您提供全方位的生信分析支持,助您轻松应对科研挑战!
)
定制生信分析
云服务器租赁
加好友
备注“99”
领取试用

题目:综合分析显示 NLRC4 是脓毒症发病机制中的潜在生物标志物
期刊:Genes & Immunity
影响因子:5.0
发表日期: 2024年8月
后台回复“
666
”获取原文献,编号20240924
研究背景
脓毒症是一种对感染的失调宿主反应,对全球公共卫生构成严重威胁,每年约有1900万人受影响,导致多达500万人死亡。因为脓毒症的早期诊断具有挑战性,因此迫切需要新型诊断方法。这篇文章旨在通过RNA测序等技术进行全面分析,突出利用基因本体(GO)分析、加权基因共表达网络分析(WGCNA)、蛋白质相互作用分析等技术,鉴定NLRC4作为脓毒症的潜在生物标志物。除此之外还探讨了单细胞RNA测序数据,发现NLRC4在脓毒症中表达水平显著增加。通过对外周血中NLRC4的鉴定,为脓毒症的诊断和治疗提供了有价值的见解。
数据来源
|
数据集
|
数据来源
|
样本数量
|
用途
|
|
GSE134347
|
GEO数据库
|
298
|
WGCNA,PPI网络分析,免疫浸润量化
|
|
GSE65682
|
GEO数据库
|
760
|
基因表达验证
|
|
GSE28750
|
GEO数据库
|
41
|
基因表达验证
|
|
GSE167363
|
GEO数据库
|
12
|
单细胞RNA测序
|
|
验证队列
|
台州市恩泽医疗中心
|
80
|
基因表达验证
|
研究思路
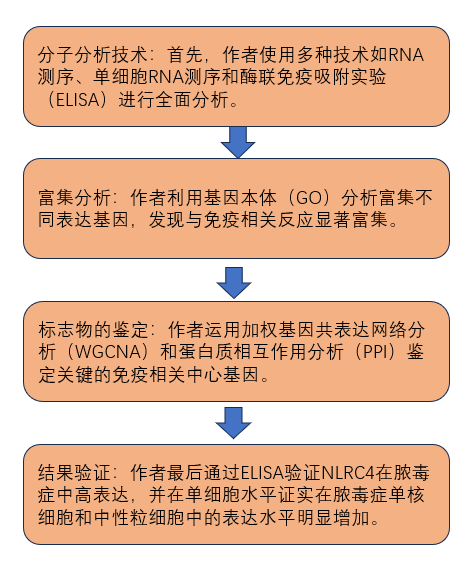
主要结果
1.数据收集
作者从
基因表达测序数据库GEO
中下载了GSE134347、GSE65682和GSE28750的基因表达数据,而验证队列包括了40名脓毒症患者和40名健康个体。除此之外,作者通过
单细胞RNA测序和10x Genomics技术
,从GSE167363数据集中获取了10个脓毒症样本,其中包括5例新诊断的脓毒症患者、5例在诊断后6小时收集的样本,以及2例健康个体的样本(表1)。
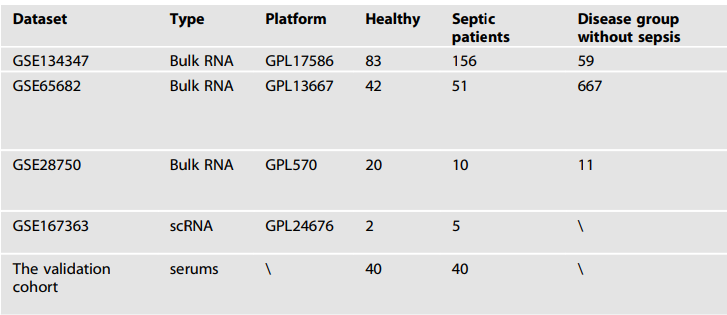
表1:相关数据集
2.差异表达分析
作者使用GSE134347数据集,包括83个健康样本、59个非感染样本和156个脓毒症样本,进行了
差异表达(DEGs)分析
。结果显示,在脓毒症样本中与健康对照组相比,共有2843个DEGs,其中1204个上调基因和1639个下调基因。此外,在脓毒症样本与非感染组之间鉴定了172个DEGs,包括114个上调基因和58个下调基因。
通过Venn图确定了这两个基因集之间的重叠DEGs,发现有161个与脓毒症相关的DEGs(图1)。
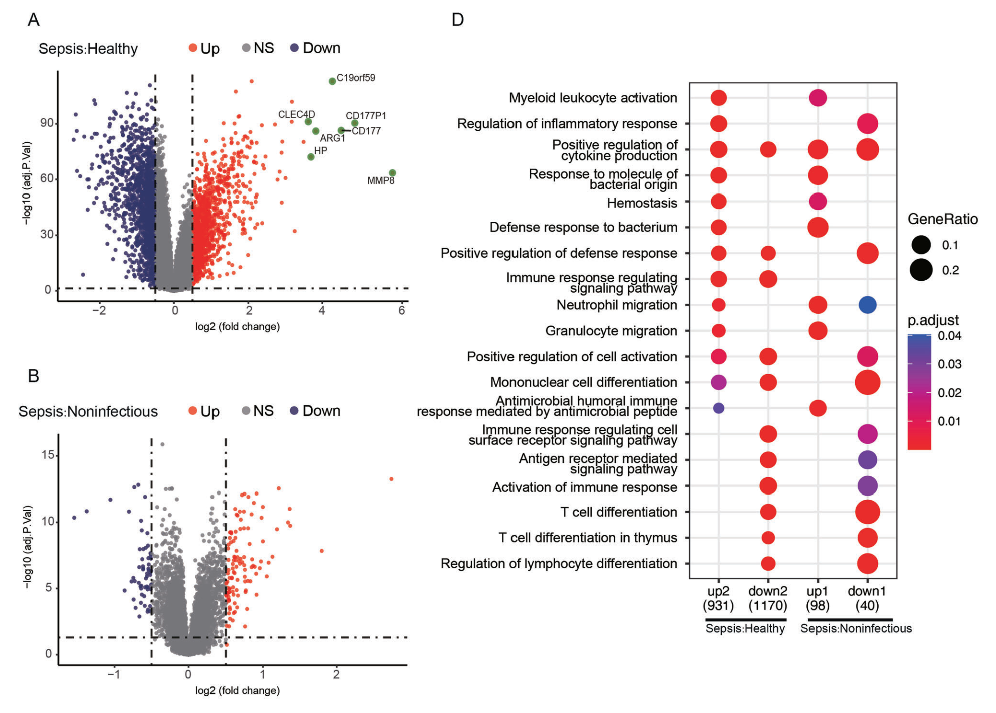
图2:脓毒症、健康和非感染性样本之间差异表达基因(DEGs)的鉴定
3.GO富集与共识聚类分析
为了探究样本中鉴定的DEGs的潜在功能,作者进行了
GO富集分析
。结果显示,在脓毒症中上调基因主要与髓样白细胞激活、炎症反应调节、对细菌的防御反应以及对细菌来源分子的反应相关,
下调基因与单核细胞分化、免疫反应激活和T细胞分化相关(图2C)
。为针对免疫相关基因进行了进一步的研究,通过
Immprot和InnateDB数据库
的交集与GSE134347数据集进行了
共识聚类分析
。结果显示,基于免疫基因表达,所有样本可分为两个簇,其中簇2主要由脓毒症和非感染组组成,
表明健康个体中免疫相关基因的激活状态可以区分脓毒症和非感染组与健康个体(图2E)。
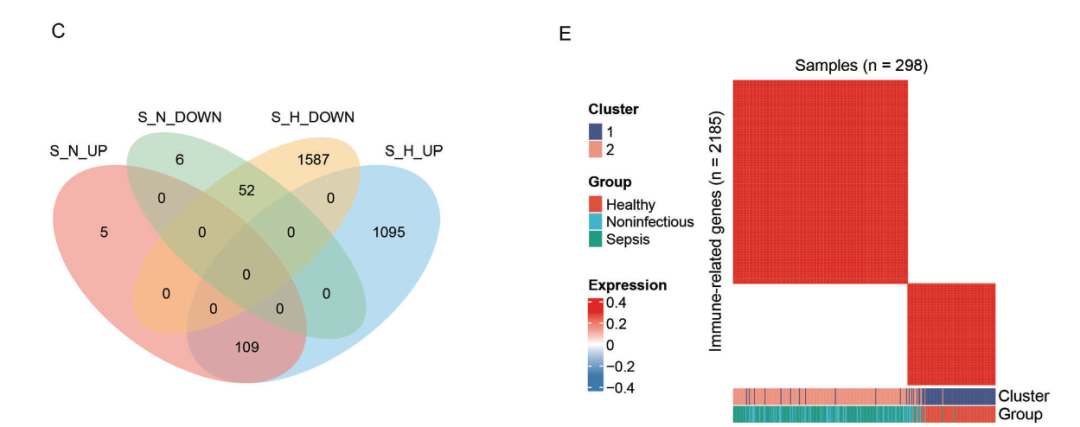
图2:失调基因维恩图与聚类矩阵。
4.WGCNA和蛋白质相互作用(PPI)网络分析
为了鉴定与脓毒症相关的关键免疫相关枢纽基因,作者使用了
WGCNA分析
GSE134347数据集中的免疫相关基因,以识别与脓毒症相关的关键模块。
结果显示,棕色模块与脓毒症之间存在强烈正相关,而蓝色模块表现出高度负相关(图3C)。
随后作者利用棕色模块的基因进行
蛋白质相互作用(PPI)网络分析
,在WGCNA分析中确定了与脓毒症高度相关的基因后,构建了包含这些基因的PPI网络。
结果发现MAP2K6和NLRC4是与脓毒症高度相关的两个免疫相关基因(图3E)。
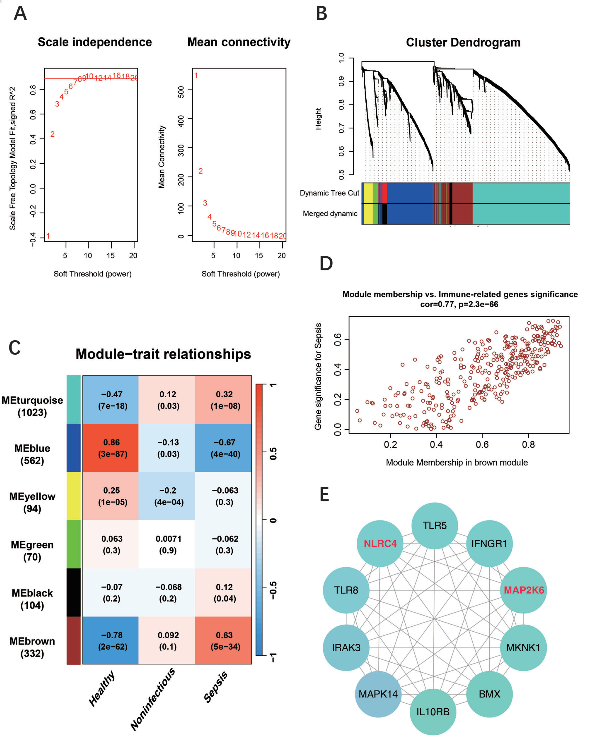
图3:使用WGCNA和PPI网络鉴定与败血症相关的关键免疫相关中心基因
5.
ROC曲线分析与ELISA验证
为了进一步调查了在脓毒症中选择的关键免疫相关基因的表达模式,作者进行了
ROC曲线分析
,评估NLRC4和MAP2K6作为脓毒症生物标志物的诊断潜力。结果表明,
NLRC4和MAP2K6在将脓毒症与健康或非感染个体区分开具有较高的AUC值,暗示它们作为脓毒症诊断生物标志物的潜力(图4C-D)。
通过
ELISA检测
血清样本,发现NLRC4在脓毒症患者的外周血中浓度显著高于健康受试者,而MAP2K6则没有显示出统计学差异(图4E)。
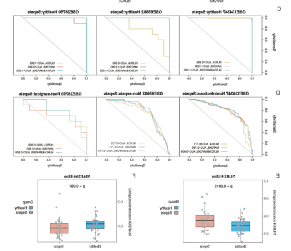
图4:ROC曲线与ELISA结果图
6.免疫浸润分析
作者利用R中的
CIBERSORT算法
计算了选择基因MAP2K6和NLRC4与各种免疫细胞类型之间的Spearman相关系数和p值。分析结果显示,在巨噬细胞M0、中性粒细胞、静止NK细胞、CD4记忆性静止T细胞和CD8 T细胞的浸润中存在显著差异(图5)。
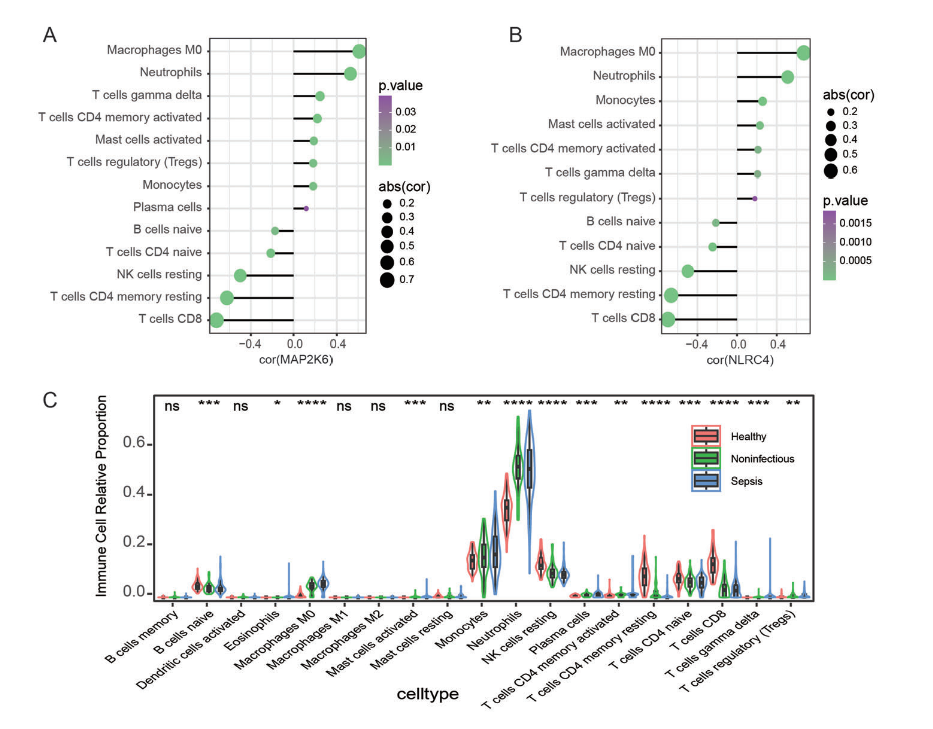
图5:CIBERSORT分析结果图










