
1. IOBR的主要分析流程
包含6大
模块
:
①数据预处理;②微环境解析和signature score计算;③微环境交互作用分析;④微环境与基因组的交互分析;⑤数据统计分析和可视化;⑥微环境模型构建;
该工具提供了丰富的微环境解析、微环境亚型鉴定、基因集评分计算方法、个性化的gene signature构建、scRNAseq的bulkseq验证、微环境相关基因变异、统计分析和可视化方法。
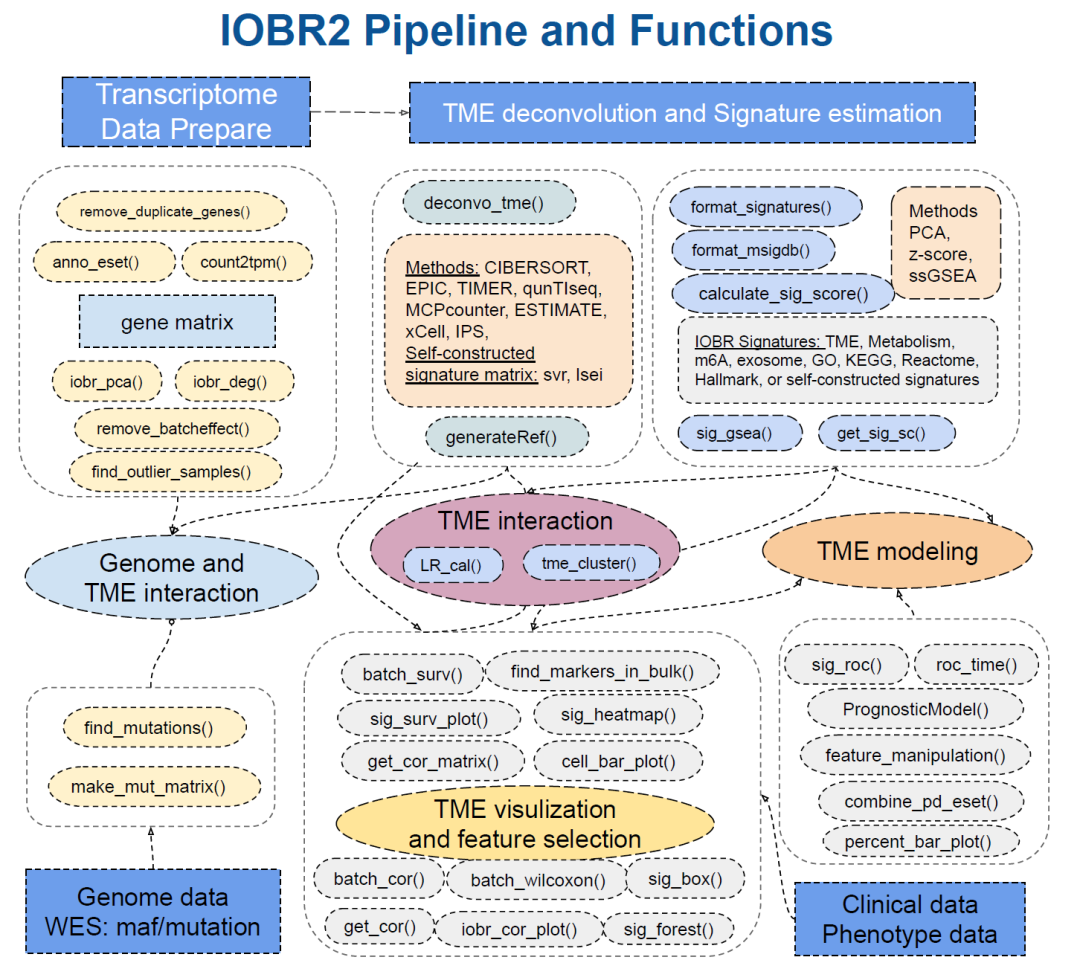
2. IOBR的函数解读
(1)数据准备
:
基因表达数据的注释和转换
-
count2tpm()
:将基因表达计数数据转换为每百万转录本(TPM)值。该函数支持“Ensembl”、“Entrez”或“Symbol”类型的基因ID,允许在线连接到bioMart数据库或本地数据集(由source参数指定)检索基因长度信息。
-
anno_eset()
:使用提供的注释数据,为 ExpressionSet 对象 (eset)注释基因 symbol。它仅保留具有注释数据中具有匹配标识符的探针的行。该函数根据指定的方法处理重复项。输出是注释和清理后的表达集。
-
remove_duplicate_genes()
:从基因表达数据中删除重复的基因 symbol。基于
基因 symbol的
平均值(如果方法设置为“mean”)或标准偏差值(如果方法设置为“sd”)来保留基因。
-
mouse2human_eset()
:将表达集的小鼠基因转化为人类基因。该函数的目的是为了将小鼠的表达矩阵转化成以人的基因symbol为行名的表达矩阵,然后用于肿瘤微环境的解析。
-
find_outlier_samples()
:分析基因表达数据并基于连通性分析(connectivity analysis)识别潜在的异常样本。通过利用“WGCNA”包,该函数计算每个样本的归一化邻接性和连通性 z 分数。它还提供多个参数来自定义分析和可视化。
-
remove_batcheffect()
:从给定的表达数据集中移除批次效应,并使用主成分分析 (PCA) 可视化校正后的数据。它接受三个表达数据集作为输入,并使用“sva::ComBat”或“sva::ComBat_seq”方法进行批次效应校正。然后,该函数生成 PCA 图以比较校正前后的数据。
(2)TME反卷积模块
:
集成多种算法来解码免疫微环境
-
deconvo_tme()
:通过各种反卷积方法,基于bulk RNAseq、微阵列数据或单细胞 RNAseq 数据,解码TME浸润模式。目前支持的方法包括
“CIBERSORT”、 “MCPcounter”、 “EPIC”、 “xCell”、 “IPS”、 “estimate”、“quanTIseq”、“TIMER”、“SVR”和“lsei”。
-
generateRef()
:生成新的基因参考数据,用于特定特征的反卷积,例如浸润细胞,利用不同的方法来识别差异表达基因(DEG)。该函数支持“limma”和“DESeq2”方法。生成的基因参考数据可用于带有“svr”和“lsei”算法的 deconvo_tme()。
-
generateRef_seurat()
:采用 Seurat 对象“sce”和附加参数来执行生成参考基因表达数据的各种操作。它允许指定细胞类型、比例、测定、预处理选项和统计测试参数。生成的基因参考数据可用于
deconvo_tme(),与
“svr”和“lsei”算法一起使用。
(3)signature模块
:
计算signature分数,估计表型相关特征和相应基因,并评估来自单细胞RNA测序数据生成的特征
-
calculate_sig_score()
:计算内置于IOBR包的特征基因集分数,涉及TME相关、肿瘤代谢和肿瘤内在特征。支持的签名分数计算方法包括“PCA”、“ssGSEA”、“z-score” ”,以及“Integration”。
-
feature_manipulation()
:对特征进行预处理,包括细胞分数和源于多组学数据的特征,用于后续分析和模型构建。预处理包括:删除缺失值、离群值、非数值和无显著方差的变量。
-
format_signatures()
:通过输入一个特征名称作为相应基因集列名的数据框,为calculate_sig_score()函数
生成
对象,并返回一个包含多个特征基因集的特征列表,用于计算多个特征分数。
-
format_msigdb()
:将gmt格式的特征基因集数据转换为calculate_sig_score()函数的对象,该数据不包含在IOBR的特征集合中,可以在MSgiDB网站上下载。
-
sig_gsea()
:
根据差异基因表达数据
进行基因集富集分析(GSEA),以识别重要的基因集。该函数使用 fgsea 包进行 GSEA,并以表格和图形的形式提供可视化和结果。它支持使用用户自定义的基因集或使用 MSigDB 中的预定义基因集。
-
get_sig_sc()
:从单细胞差异分析中提取每个细胞类型排名靠前的基因,作为相应细胞类型的特征基因集,为
calculate_sig_score()函数生成对象
。输入是一个包含
假定marker的排名列表以及相关统计数据(p 值、ROC 分数等)的
矩阵。
(4)批量分析和可视化:
批量生存分析和相关性分析等其他批量统计分析
-
batch_surv()
:执行批量生存分析。它根据包含时间相关信息的给定数据,计算指定变量的风险比和置信区间。
-
subgroup_survival()
:从亚组分析的coxph对象中提取风险比和置信区间。
-
batch_cor()
:使用皮尔逊相关系数或斯皮尔曼等级相关系数,对两个连续变量之间的相关性进行批量分析。
-
batch_wilcoxon()
:对给定数据集执行 Wilcoxon 秩和检验,以比较两组之间指定特征的分布。它计算 p 值,并根据 p 值对显著特征进行排名。它返回一个数据框,其中包含特征名称、p 值、调整后的 p 值、p 值的对数,以及基于 p 值范围的星级评定。
-
batch_pcc()
:提供一种批量处理方法,在控制第三个变量的情况下,计算特征与其他特征之间的偏相关系数。
-
iobr_cor_plot()
:对“sig_group”特征的批量相关性分析结果的可视化。还支持可视化特征或表型与目标特征中基因集的表达之间的相关性。
-
cell_bar_plot()
:TME细胞分数的批量可视化,支持输入“CIBERSORT”、“EPIC”和“quanTIseq”方法的反卷积结果,以进一步比较一个样本内或不同样本之间的TME细胞分布。
-
iobr_pca()
:执行主成分分析(PCA),在保持大部分原始方差的同时降低数据的维数,并在散点图上可视化PCA结果。
-
iobr_deg()
:使用DESeq2或limma方法对基因表达数据进行差异表达分析。它过滤低计数数据,计算倍数变化和调整后的 p 值,并根据指定的截止值识别 DEG。
-
get_cor()
:计算并可视化数据集中两个变量之间的相关性。它提供了缩放数据、处理缺失值和合并额外数据的选项。该函数支持多种相关性分析方法。它能根据亚组类型/类别生成散点图并添加回归线,此外还提供了多种自定义可视化选项。
-
get_cor_matrix()
:计算并可视化数据集中两组变量之间的相关矩阵。它能够灵活地定义相关方法、处理缺失值和合并附加数据。该函数支持各种相关性分析方法,例如“Pearson correlation”,并在可自定义的图中显示相关结果。
-
roc_time()
:生成随时间变化的接收者操作特性(ROC)图,以评估生存分析中一个或多个变量的预测性能。它计算每个指定时间点和变量组合的曲线下面积 (AUC),并创建一个带有相应 AUC 值注释的多线 ROC 图。
-
sig_box()
:生成带有可选统计比较的箱线图。它接受各种参数(例如数据、特征、变量等)来自定义绘图。它可用于可视化和分析 Seurat 对象或任何其他数据框中的数据。
-
sig_heatmap()
:根据输入数据、分组变量和可选条件生成热图。该功能允许自定义各种参数,例如调色板选择、缩放、颜色框、绘图尺寸等。它以简洁且信息丰富的方式灵活地可视化变量和组之间的关系。
-
sig_forest()
:创建森林图,用于可视化“batch_surv”生成的生存分析结果,可通过参数设置需要展示的变量数量。
-
sig_roc()
:在单个图中绘制多个 ROC 曲线,便于比较不同变量预测二元响应方面的能力。
-
sig_surv_plo
t():为给定的基因、基因评分和细胞浸润评分生成多个 Kaplan-Meier (KM) 生存图。它允许进行详细的定制,并且结构化以处理生存分析的各个方面。
-
find_markers_in_bulk()
:从给定的基因表达数据和元信息中查找相关结果。它利用“Seurat”包中的FindAllMarkers()函数来识别给定数据中多个组的显著变量。支持的比较方法包含 “bootstrap”、“delong”和“venkatraman”。
(5)signature相关突变模块
:
识别和分析与目标特征相关的突变
-
make_mut_matrix()
:以合适的方式将MAF格式的突变数据(包含基因ID列和相应的基因改变,包括SNP、indel和移码)转换为突变矩阵,以进一步研究与特征相关的突变。
-
find_mutations()
:识别与不同表型或特征相关的突变。该函数进行 Cuzick 检验、Wilcoxon 检验,或两者同时进行(当方法设置为“multi”时)。它为通过这些统计检验识别的最显著的基因生成箱线图,并创建 oncoprints ,以图形方式展示样本间的突变景观。










