果子荐读
不一会的功夫,春卷的CRISPR/cas9基因编辑推文第一部分已经完成,她用5篇帖子完成了从零开始到选择有效sgRNA的全部过程。
接下来就是真正的开始。
在文末,她还有一段真诚的总结。
我们公众号目前有三个实验专栏,分别是高师姐,豆子还有春卷,当我们在写作的时候,并不是居高临下地要求大家跟着做,而是提供新的思路,开拓眼界。
一个研究生,只要做过一段时间实验,就会慢慢形成自己的心得体会,甚至最终变成自己的护城河。极端的师兄甚至会告诉自己的师弟师妹,细胞吹打,108下,不能多也不能少,要不然细胞就养不好。
我们的三个专栏就是来击穿这个迷信的。
高师姐并没有跟豆子学习WB,你甚至可以看到面对同样一个实验,他们的心得完全不一样,但是,不妨碍他们自己都在发10分的paper
他们各自的体系是自洽的,跟着任何一位都可以完全掌握科研技能。
而那些心得不过是手熟后的自然而然的产物,在这之前他们哪一个不是投入了大量的时间呢。
我们有很多小伙伴,总是犹豫自己是否该学一门新的语言,比如编程,有R,有Python,有Perl,有Juli,
选择起来很困难,
假如你听了我的话
如果临床医生只学一门语言,那就是R语言。
那么还会面临很多问题,我该学习R语言基础,还是直接从tidyverse开始,画图是用base plot打好基础还是直接起手就是ggplot2?
焦虑年年有,选择的时候特别多。
但,所有的这些焦虑都会在你写完1000行代码后荡然无存。
当用shell完成了dplyr的操作后,所有买过的linux书突然变成了立体的,我知道了取舍;
当用Python完成了一次data wrangling后,我发现我手上90%的Python资料都可以扔掉,为什么之前不觉得呢。
当完成了一次爬虫,html,css,JavaScript,代理池,代理cookie,这些看起来无比高深的知识也能接受了。
这就是我今年感受最深的地方,
当你想要学习一项新技能的时候,趁着心是热的,那怕只有三分钟,你赶紧去学,要不然一会就要改变主意的。
而改变主意最糟糕的地方在于,你并没有死心,当下次被某个讲座再次燃起热情的时候,你又开始了无休无止的资料收集工作,收集完成后仿佛有了世界,然而却从不开始真正的学习。
以下是正文:
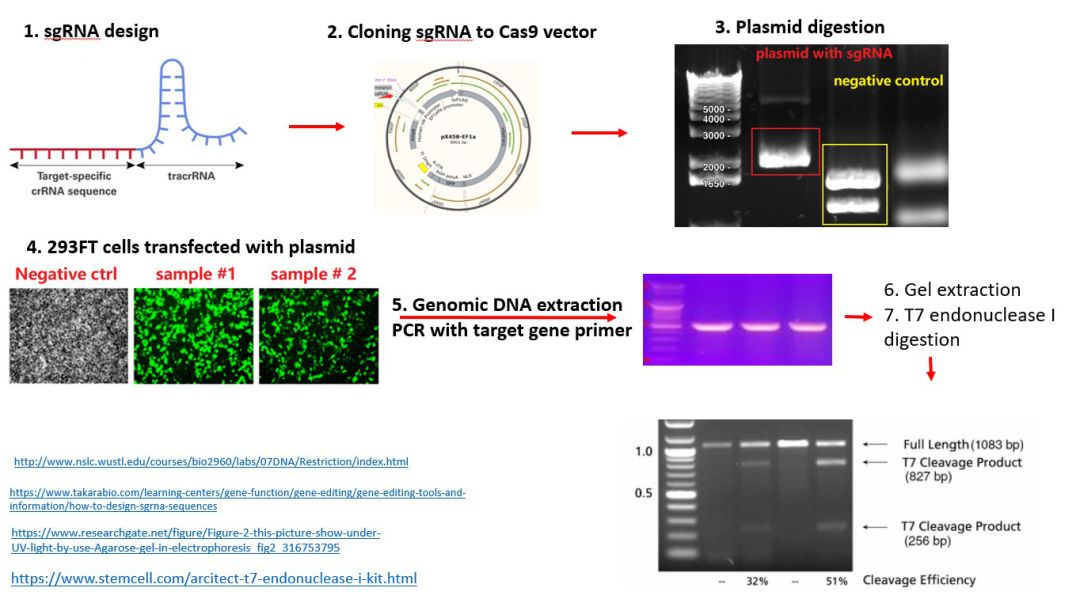
大家都知道CRISPR-Cas9有脱靶效应,并且每个sgRNA的效率都不一样的,因此在做正式实验之前,我们要验证sgRNA的效率,之后优中选优。还是那句话,同样的目的有很多不同的方案,我了解到的有:
-
使用sgRNA靶点筛选试剂盒,优点快速、工作量小且可以大批量进行;
-
将sgRNA-pSpCas9转入293细胞中,提取293细胞基因组DNA,PCR扩增目的DNA片段后,使用T7核酸内切酶,验证sgRNA切割效率;
欢迎大家补充,互相交流,提供更方便快捷的方案,由于我们实验室购买试剂盒时间长,相对困难,且sgRNA的切割效率的验证还是活细胞内效率最为准确。经过课题组师兄DZ的优化,一般我们针对一个基因的简单敲除,设计2个sgRNA即可,并且选择方案2进行验证。
接前期推文,我们做了一手漂亮的质粒转染后,
如何确定sgRNA切割活性,这条sgRNA到底还要不要,就看今天了
。
实验正文
1. 实验准备
2. 实验步骤
(1)293细胞转染
在转染前一天,将293细胞以单细胞分散状态种至6孔板中,组别设计为:
293未转染组(negative control),293转染组
。转染当天293细胞融合度为30~40%,将2.0~2.5 μg pSpCas9_sgRNA质粒使用lipofectamine 3000按照常规条件转染(具体细节请参照上期文章),72 h内进行下一步实验。
注意事项:由于293细胞贴壁不牢,很容易被吹落,因此在滴加转染试剂的时候要十分轻柔,换液时要更加小心沿壁加入,并且培养基要提前预温。
(2)提取293细胞基因组DNA
这里我们使用的是天根公司的基因组DNA提取试剂盒,严格按照试剂盒要求即可。
实验细节调整:
原protocol可能需要消化细胞后再加裂解液,在这里由于293细胞非常容易脱落,因此我选择不消化细胞,弃去上清,直接使用孔种的培养基吹落细胞,移入1.5 ml EP管离心去除上清后,直接加入裂解液吹打,之后步骤同试剂盒要求一样。
(3)使用高保真DNA聚合酶完成PCR实验
一定要使用高保真酶的原因是,T7 EI内切酶的原理是识别突变位点,所以如果使用的DNA聚合酶不够保真,在PCR过程扩增错误,引入突变,则会影响后续结果。
在这里需要提醒的是,任何DNA聚合酶的使用,一定要严格查阅相应说明书,确定实验条件
。我们使用的是NEB公司的Q5高保真DNA聚合酶,根据说明书指示,Q5 DNA聚合酶做的PCR实验,其primer的退火温度和普通我们使用的不一样,需要使用NEB公司的Tm calculator工具计算得出。
(4)胶回收PCR扩增出的目的序列
胶回收的原则就是,量大质优,严格按照说明书操作即可。
(5)T7 Endonuclease I 酶切实验
配置实验反应
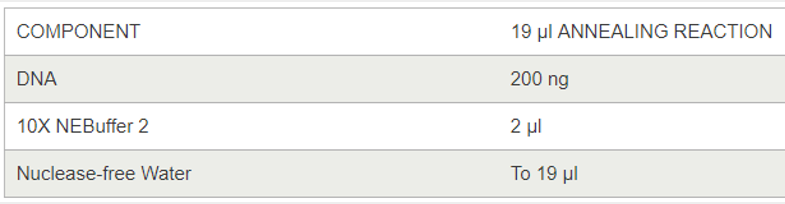
按照以下条件annealing
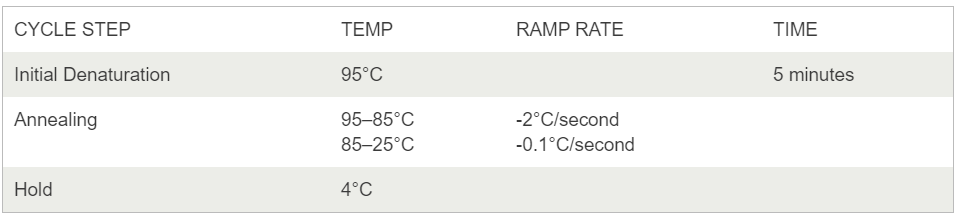
设置酶切反应后,37 ℃孵育15分钟

(6)琼脂糖凝胶电泳,预测分子量大小<1 kb,使用 1.5~2%琼脂糖;分子量大小为1~10 kb大小时,选用1-1.2%
(7)结果分析示例
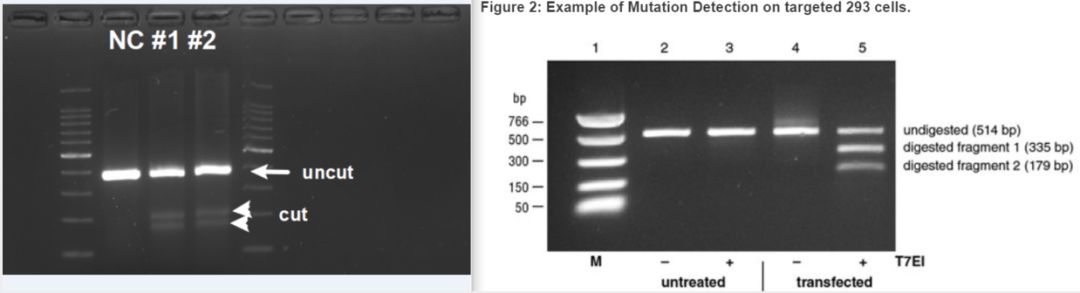
对于左图
(1)可以看到negative control组中,T7酶并没有将DNA切断,可以说明没有出现基因错配的情况;
(2)而分别用了sgRNA 1 和sgRNA 2的两组,均出现了DNA被切割的现象,且切割后的片段长度加起来,恰好等于uncut line。
以上结果说明,在未转染sgRNA-Cas9组(NC)中,DNA保持了原有的正确序列,转染sgRNA-Cas9质粒后的细胞,基因已经被编辑。sgRNA 1和2均可切割,且量条sgRNA的效率差不多,随手选一条即可。
右图是更加标准的做法,左图少了blank control 组(不加T7 酶的组)
。
我是小结
CRISPR-Cas9基因编辑的直接敲除系列,今天写到这里算是完成了预实验部分。这个时候可以把整个方案的流程放在这里权当总结,并提示之后实验方向方向:
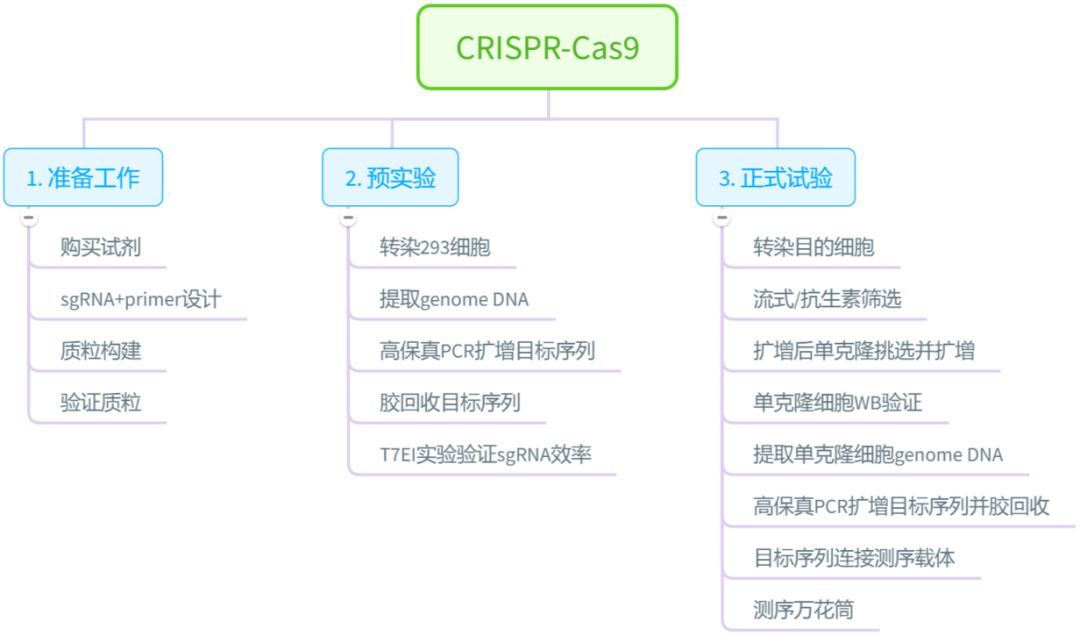
准备工作和预实验要至少得到以下简化的实验结果:
作者心声
在结尾我想分享一下自己的小小感想:首先感谢果子老师给我这个机会,发布我的系列实验文章,平时写的实验记录和经验分享,受众都是我自己,因此无论好坏,有无含糊,都没有那么在意。当初果子老师跟我提这事儿的时候我犹豫了,因为我们每个人都要对自己的言论和行为负责,一旦放到公众号上,这就说明我要对自己写过的东西负责,应尽自己最大努力去保证我所发布的内容没有误导读者,同时也要保证自己写的不浪费读者的时间,要有干货不掺水。因为有这样的原则在,写教程最大的受益者是我自己,因为我写得有多细,就代表我学得有多少,读者不一定会复刻我的所有实验,但我却能复盘我的每一次实验思路,读者的评论或建议,这我打开自己新思路的机会,所以也要感谢大家。最后,再次感谢我的师兄DZ,是他不厌其烦的解答我的问题,无保留的传授我实验方案,感恩。
下周三见~





