主要观点总结
该文章报道了一种基于硫胺素依赖酶(ThDP)的光生物协同催化体系,能够实现三组分立体选择性交叉偶联反应。该反应利用简单易得的原料产生自由基,通过醛分子产生酰基自由基,α-溴羰基化合物产生亲电自由基,烯烃作为自由基受体,直接合成含有丰富立体结构的有机酮。通过酶的定向进化,反应效率显著提高,具有优异的立体选择性控制、温和反应条件和广泛的官能团兼容性。
关键观点总结
关键观点1: 研究背景
多组分反应在酶催化中较为罕见,尤其是涉及多个自由基中间体的反应。近期发展的自由基分选策略难以控制立体选择性。
关键观点2: 主要研究成果
黄小强教授和王斌举教授等报道了利用焦磷酸硫胺素依赖酶进行定向进化,结合光催化剂,实现了三组分立体选择性交叉偶联。该反应展现出完美的立体选择性,并在一系列测试中表现出高选择性和兼容性。
关键观点3: 反应设计和机制
作者基于硫胺素依赖酶(ThDP)开发了一种光/生物协同催化体系,通过简单原料产生自由基。反应机理揭示了光-酶协同催化体系能够精确形成三个自由基,构成新型酶催化反应。
关键观点4: 反应优化和兼容性
通过酶的定向进化,反应效率显著提高。该反应具有广泛的官能团兼容性,能够合成具有复杂结构的分子。
关键观点5: 研究影响
该研究为酶催化多组分反应提供了新的思路和方法,有望在有机合成和药物研发等领域得到广泛应用。
正文

多组分反应(Multicomponent reaction)是三个反应物或者更多反应物结合构筑一个产物的反应,多组分反应能够快速合成结构复杂的化合物,但是对于酶催化剂,多组分反应仍然非常罕见。这是因为酶的催化活性位点通常无法支持多个反应物,特别是对于含有多个自由基中间体的反应。最近,人们发展了自由基分选(radical sorting)策略能够对多个反应物参与的自由基转化反应,但是这种反应难以控制立体选择性。
有鉴于此,
南京大学黄小强教授、
厦门大学王斌举教授等
报道焦磷酸硫胺素依赖酶进行定向进化,并且结合光催化剂,实现了三组分立体选择性交叉偶联。
这个反应能够将容易获取的三个反应物(醛、α-溴羰基化合物、烯烃)转化为立体结构的酮。
反应机理揭示光-酶协同催化体系能够精确形成三个自由基,构成了新型酶催化反应。这项反应表现了完美的立体选择性,测试的33个反应中,25个反应的立体选择性≥97%。
硫胺素依赖酶(ThDP)是自然界中非常常见的两电子形成化学键和断裂化学键。同时,最近人们开发的化学催化自由基分选机理拓展了自由基交叉偶联反应,但是目前此类过程仍无法进行立体选择性。
作者基于硫胺素依赖酶(ThDP)开发了一种光/生物协同催化体系,通过简单易得原料产生自由基。通过醛分子产生酰基自由基,α-溴羰基化合物产生亲电自由基,烯烃作为自由基的受体。利用重组的三组分自由基酶直接合成含有丰富立体结构的有机酮。
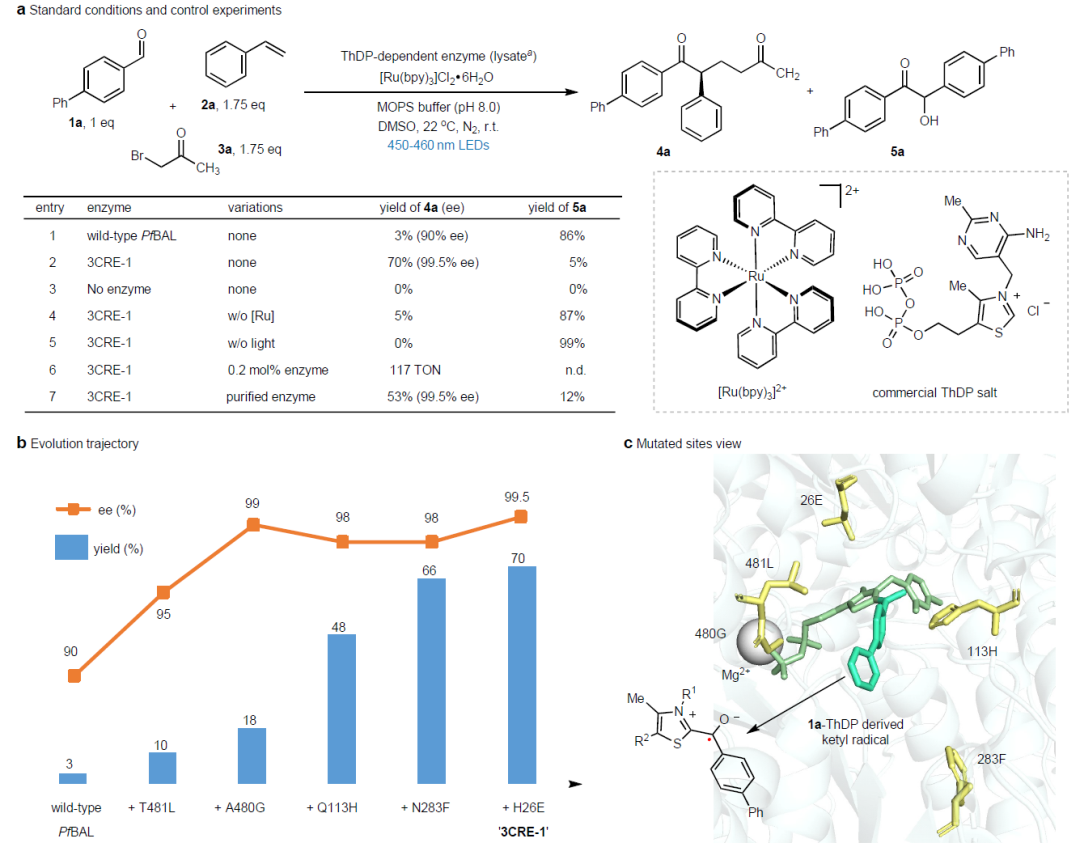 图2.
反应条件优化
以[1,1′-联苯]-4-甲醛(
1a
)分子产生酰基、苯乙烯(
2a
)作为烯烃自由基受体、1-溴-2-丙酮(
3a
)产生亲电自由基。使用Ru(bpy)
3
Cl
2
作为光催化剂,蓝光(波长450-460nm)LED作为光源,筛选硫胺素依赖酶ThDP,发现
Pseudomonas fluorescens
(
Pf
BAL)的苯甲醛裂解酶能够催化三组分反应,但是产率仅为3%,立体选择性为90% ee(Entry)。这种野生型
Pf
BAL主要面临的困难是反应生成偶姻缩合反应产物
5a
。
酶的定向进化。通过半理性迭代定点突变策略对野生
Pf
BAL酶进行结构进化,得到最优结构的变体3CRE-1,能够以70%的收率和99.5%的立体选择性生成
4a
,反应的苯偶姻缩合副反应选择性得到非常好的抑制(5%
5a
),(Entry 2)。分别通过位点饱和突变、分子动力学模拟辅助筛选的多轮过程得到3CRE-1变体。控制实验验证了ThDP辅酶、光催化剂、可见光是该反应必不可少的挑战。通过酶的定向进化,催化反应效率显著提高,TON达到117。
酰基自由基反应物兼容性。对一系列对位芳基/杂芳基取代基的醛,能够实现优异的立体选择性(≥99% ee)(
4a-f
)。其中对位杂芳基酮(
4c-f
)是非常有用的过渡金属催化C-H键官能团化反应物,同时具有弱配位点和强配位点。含有富电子或者缺电子取代基的芳基醛(
4g-k
)兼容,对邻位修饰氟官能团的氟苯甲醛(
4l
)、苯并噻吩醛(
4m
)有良好的兼容性。
烯烃兼容性。该反应对许多常见烯烃有着良好的反应性,能够合成立体结构1,5-二酮化合物,对各式各样的电子结构(
4n-q
)以及不同位点取代基(
4q-s
)兼容。这种光生物催化体系兼容许多杂环烯烃化合物,包括噻吩(
4t
)、呋喃(
4u
)、吡啶(
4v
)等官能团,能够将共轭二烯烃转化为1,7-二酮(
4w
)。反应兼容没有活化的脂肪族烯烃(
4x
),表明该反应的广泛应用前景。
该反应兼容许多官能团。兼容的官能团包括酮(
4y-aa
)、酯(
4ab
,
4ae
)、腈基(
4ac
)。使用市售二氟甲基化试剂
3g,能够生成立体结构富集的二氟酮(4ad
),反应兼容重氮化合物(
4ae
)。消旋的α-卤酮能够生成不同立体比例的二酮(
4af
,
4ag
)。
反应可进行放大量合成,以63%的产率生成
4a,以61%的产率生成4b,以36%的产率生成4ab
,且放大量没有影响产物立体选择性。
该反应具有优异的立体选择性控制,温和反应条件,对许多官能团容忍,这种新型生物催化反应显著拓展了分子的复杂性。
提出了光催化-酶催化协同催化反应。首先硫胺素依赖酶(ThDP)的3CRE变体和
1
醛分子反应生成Breslow中间体(
Int. A
)。随后被[Ru]*光催化剂单电子氧化-脱氢生成酰基自由基
Int. B,同时还原的[Ru]
·
-
将自由基前体
3
进行单电子还原,生成亲电自由基
Int. C
。
随后
Int. C
对烯烃分子2自由基加成,生成前手性亲核性自由基
Int. D
,最后在酶催化活性位点发生化学选择性和立体选择性自由基交叉偶联反应,生成立体结构的有机酮。
通过实验和理论计算验证机理。通过Stern-Volmer还原淬灭反应,说明在只有醛(
1a
)和酶的体系,发生还原淬灭[Ru]*。计算Breslow中间体(
Int. A
)对[Ru]*的电子转移,发现电子转移放热46.4kcal mol
-1
,在热力学上容易发生。相比的氧化淬灭反应为吸热过程(吸热21.8kcal mol
-1
),因此
确立光催化反应的还原淬灭过程
。
通过分子对接和分子动力学模拟,验证[Ru]能够通过π-π相互作用,结合在酶催化活性位点的F283基团附近。因此
[Ru]和
Int. A
之间距离靠近,能够高效率的转移电子和中间体,促进三组分自由基反应的高效率
。高效率的淬灭反应同样验证这个结论。
分子动力学模拟结果表明H113基团作为碱,对
Int. A
脱质子生成自由基阳离子(
Int. A
·+
),促进转化为
Int. B
。通过量子力学/分子动力学(QM/MM)计算验证
Int. A
·+
容易对H113基团转移质子(能垒仅为2.1kcal mol
-1
,反应放热4.5kcal mol
-1
)。
验证[Ru]
·-
进行单电子还原能够产生
Int. C
。该反应为放热过程(3.9kcal mol
-1
),通过室温DMPO捕获实验进行ESR测试,验证生成DMPO/
Int. C
加合物。在靠近酶催化口袋或者酶催化口袋内部生成
Int. B
和
Int. C
。通过控制实验验证,
生成C(sp
2
)-C(sp
3
)的关键步骤在酶活性活性位点进行,因此能够得以保证酶催化剂能够控制反应产物的立体选择性
。
Xing, Z., Liu, F., Feng, J.
et al.
Synergistic photobiocatalysis for enantioselective triple radical sorting.
Nature
(2024)
DOI: 10.1038/s41586-024-08399-5
https://www.nature.com/articles/s41586-024-08399-5
图2.
反应条件优化
以[1,1′-联苯]-4-甲醛(
1a
)分子产生酰基、苯乙烯(
2a
)作为烯烃自由基受体、1-溴-2-丙酮(
3a
)产生亲电自由基。使用Ru(bpy)
3
Cl
2
作为光催化剂,蓝光(波长450-460nm)LED作为光源,筛选硫胺素依赖酶ThDP,发现
Pseudomonas fluorescens
(
Pf
BAL)的苯甲醛裂解酶能够催化三组分反应,但是产率仅为3%,立体选择性为90% ee(Entry)。这种野生型
Pf
BAL主要面临的困难是反应生成偶姻缩合反应产物
5a
。
酶的定向进化。通过半理性迭代定点突变策略对野生
Pf
BAL酶进行结构进化,得到最优结构的变体3CRE-1,能够以70%的收率和99.5%的立体选择性生成
4a
,反应的苯偶姻缩合副反应选择性得到非常好的抑制(5%
5a
),(Entry 2)。分别通过位点饱和突变、分子动力学模拟辅助筛选的多轮过程得到3CRE-1变体。控制实验验证了ThDP辅酶、光催化剂、可见光是该反应必不可少的挑战。通过酶的定向进化,催化反应效率显著提高,TON达到117。
酰基自由基反应物兼容性。对一系列对位芳基/杂芳基取代基的醛,能够实现优异的立体选择性(≥99% ee)(
4a-f
)。其中对位杂芳基酮(
4c-f
)是非常有用的过渡金属催化C-H键官能团化反应物,同时具有弱配位点和强配位点。含有富电子或者缺电子取代基的芳基醛(
4g-k
)兼容,对邻位修饰氟官能团的氟苯甲醛(
4l
)、苯并噻吩醛(
4m
)有良好的兼容性。
烯烃兼容性。该反应对许多常见烯烃有着良好的反应性,能够合成立体结构1,5-二酮化合物,对各式各样的电子结构(
4n-q
)以及不同位点取代基(
4q-s
)兼容。这种光生物催化体系兼容许多杂环烯烃化合物,包括噻吩(
4t
)、呋喃(
4u
)、吡啶(
4v
)等官能团,能够将共轭二烯烃转化为1,7-二酮(
4w
)。反应兼容没有活化的脂肪族烯烃(
4x
),表明该反应的广泛应用前景。
该反应兼容许多官能团。兼容的官能团包括酮(
4y-aa
)、酯(
4ab
,
4ae
)、腈基(
4ac
)。使用市售二氟甲基化试剂
3g,能够生成立体结构富集的二氟酮(4ad
),反应兼容重氮化合物(
4ae
)。消旋的α-卤酮能够生成不同立体比例的二酮(
4af
,
4ag
)。
反应可进行放大量合成,以63%的产率生成
4a,以61%的产率生成4b,以36%的产率生成4ab
,且放大量没有影响产物立体选择性。
该反应具有优异的立体选择性控制,温和反应条件,对许多官能团容忍,这种新型生物催化反应显著拓展了分子的复杂性。
提出了光催化-酶催化协同催化反应。首先硫胺素依赖酶(ThDP)的3CRE变体和
1
醛分子反应生成Breslow中间体(
Int. A
)。随后被[Ru]*光催化剂单电子氧化-脱氢生成酰基自由基
Int. B,同时还原的[Ru]
·
-
将自由基前体
3
进行单电子还原,生成亲电自由基
Int. C
。
随后
Int. C
对烯烃分子2自由基加成,生成前手性亲核性自由基
Int. D
,最后在酶催化活性位点发生化学选择性和立体选择性自由基交叉偶联反应,生成立体结构的有机酮。
通过实验和理论计算验证机理。通过Stern-Volmer还原淬灭反应,说明在只有醛(
1a
)和酶的体系,发生还原淬灭[Ru]*。计算Breslow中间体(
Int. A
)对[Ru]*的电子转移,发现电子转移放热46.4kcal mol
-1
,在热力学上容易发生。相比的氧化淬灭反应为吸热过程(吸热21.8kcal mol
-1
),因此
确立光催化反应的还原淬灭过程
。
通过分子对接和分子动力学模拟,验证[Ru]能够通过π-π相互作用,结合在酶催化活性位点的F283基团附近。因此
[Ru]和
Int. A
之间距离靠近,能够高效率的转移电子和中间体,促进三组分自由基反应的高效率
。高效率的淬灭反应同样验证这个结论。
分子动力学模拟结果表明H113基团作为碱,对
Int. A
脱质子生成自由基阳离子(
Int. A
·+
),促进转化为
Int. B
。通过量子力学/分子动力学(QM/MM)计算验证
Int. A
·+
容易对H113基团转移质子(能垒仅为2.1kcal mol
-1
,反应放热4.5kcal mol
-1
)。
验证[Ru]
·-
进行单电子还原能够产生
Int. C
。该反应为放热过程(3.9kcal mol
-1
),通过室温DMPO捕获实验进行ESR测试,验证生成DMPO/
Int. C
加合物。在靠近酶催化口袋或者酶催化口袋内部生成
Int. B
和
Int. C
。通过控制实验验证,
生成C(sp
2
)-C(sp
3
)的关键步骤在酶活性活性位点进行,因此能够得以保证酶催化剂能够控制反应产物的立体选择性
。
Xing, Z., Liu, F., Feng, J.
et al.
Synergistic photobiocatalysis for enantioselective triple radical sorting.
Nature
(2024)
DOI: 10.1038/s41586-024-08399-5
https://www.nature.com/articles/s41586-024-08399-5





