
我最近正好在学习GWAS(全基因组关联分析),这一套流程呢简单来说就是在全基因组范围内寻找变异序列,在全基因组范围内进行相关性分析,通过比较发现复杂性状的基因变异。
我学习的过程中参考的是
黄学辉教授
15年发表在NC上的文章“Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis”。里面介绍到分析过程中运用到的一些软件和方法。
原始数据的处理,变异检测(BWA+SAMtools+picard+GATK)这套流程进行SNP calling生成VCF文件我就不介绍了。
在介绍GWAS之前,有几个分析过程中产生的文件,有几个格式需要先稍微介绍一下。
变异信息存放格式之VCF
VCF是用于描述SNP,INDEL,SV的文本文件。是GATK表示遗传变异的一种文件格式。
可以分成两个部分来看:
第一部分是以
##
开头的说明文件,解释第二部分INFO列中可能要出现的一些tags和和FORMAT列中对基因型的表示。个人觉得最个注释没什么用处,我基本都是直接跳过的。
##fileformat=VCFv4.1
##FILTER=
##FORMAT=
##FORMAT=
##FORMAT=
##INFO=
##INFO=
##contig=
##contig=
##contig=
##contig=
第二部分是重点了,
正文部分
主要包括9列+N样品列
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT B73 F2-Mo17vsB73 Mo17
1 530 . C G 52.17 . AC=2;AF=0.333;AN=6;BaseQRankSum=-0.948;DP=535;Dels=0.00;FS=14.393;HaplotypeScore=11.0860;MLEAC=1;MLEAF=0.167;MQ=6.29;MQ0=396;MQRankSum=2.281;QD=0.15;ReadPosRankSum=0.530;SOR=3.223 GT:AD:DP:GQ:PL 0/1:208,3:218:13:13,0,100 0/0:176,0:177:39:0,39,297 0/1:136,2:140:31:45,0,31
1 534 . G A 32.35 . AC=1;AF=0.167;AN=6;BaseQRankSum=-0.117;DP=539;Dels=0.00;FS=10.307;HaplotypeScore=15.3371;MLEAC=1;MLEAF=0.167;MQ=6.36;MQ0=397;MQRankSum=3.126;QD=0.15;ReadPosRankSum=0.154;SOR=1.431 GT:AD:DP:GQ:PL 0/1:206,4:214:63:63,0,117 0/0:178,2:182:33:0,33,252 0/0:139,0:143:12:0,12,101
1 542 . C T 32.35 . AC=1;AF=0.167;AN=6;BaseQRankSum=-1.405;DP=534;Dels=0.00;FS=11.442;HaplotypeScore=12.8859;MLEAC=1;MLEAF=0.167;MQ=6.38;MQ0=391;MQRankSum=2.054;QD=0.15;ReadPosRankSum=-0.330;SOR=2.221 GT:AD:DP:GQ:PL 0/1:207,10:218:63:63,0,117 0/0:175,3:178:39:0,39,297 0/0:134,3:138:12:0,12,101
CHROM
和
pos
:表示变异位点相对reference的位置,比如第几条染色体的第几个碱基,如果是indel,pos是indel的第一个碱基的位置
ID
:如果call出来的SNP存在于dbsnp数据库里,就会显示相应的dbsnp里的rs编号。不然就是用“.”表示一个novel variant.
REF和ALT
:分别代表reference和alter,也就是参考基因组对应的碱基和variant的碱基。
QUAL
:表示该位点存在variant的可能性,qual值越大则variant的可能性越大。
FILTER
:过滤完了之后,FILTER一栏会有过滤记录,通过了过滤标准,那么这些好的变异位点的FILTER一栏就会注释一个
PASS
,如果没有通过过滤,就会在FILTER这一栏提示其他信息。如果这一栏是一个“.”的话,就说明没有进行过任何过滤。
IFNO
:这一列表示的是variant的详细信息。
-
GT:表示样本的基因型,对于一个二倍体生物,GT值表示的是这个样本在这个位点所携带的两个等位基因。0表示跟REF一样;1表示表示跟ALT一样;2表示第二个ALT。当只有一个ALT等位基因的时候,0/0表示纯和且跟REF一致;0/1表示杂合,两个allele一个是ALT一个是REF;1/1表示纯和且都为ALT;
-
AD:对应两个以逗号隔开的值,这两个值分别表示覆盖到REF和ALT碱基的reads数,相当于支持REF和支持ALT的测序深度。
-
DP:覆盖到这个位点的总的reads数量,相当于这个位点的深度(并不是多有的reads数量,而是大概一定质量值要求的reads数)。
-
PL:对应3个以逗号隔开的值,这三个值分别表示该位点基因型是0/0,0/1,1/1的没经过先验的标准化Phred-scaled似然值(L)。如果转换成支持该基因型概率(P)的话,由于L=-10lgP,那么P=10^(-L/10)^,因此,当L值为0时,P=10^0^=1。因此,这个值越小,支持概率就越大,也就是说是这个基因型的可能性越大。
-
GQ:表示最可能的基因型的质量值。表示的意义同QUAL。
变异信息存放格式之 tped
tped格式:是varient信息+基因型调用的文本文件
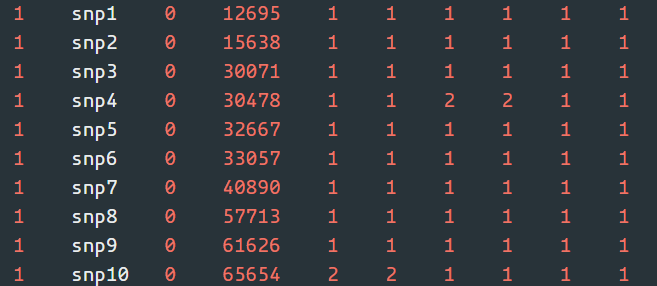
tped文件时没有header的。每一行有4+2N个条目,N代表样本数目。
后面的都表示样本列,一个二倍体的样本会有两列。两个数分别表示等位基因,可以用0、1;1、2表示。’12’修饰符表示A1等位基因编码为’1’,A2等位基因编码为’2’,而’01’则映射A1→0和A2→1。
变异信息存放格式之 tfam
tfam格式: tfam文件和tped文件相对应,tfam文件样品是纵向排列而tped(第五列开始)横向排列。
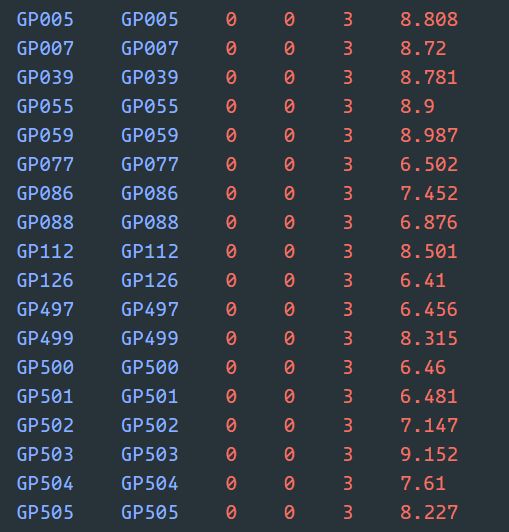
tfam文件每行包括6个条目
-
第一列是样本ID(FID)
-
第二列是样本内部ID(IID;不能为‘0’)
-
第三列父本ID(如果父本不在数据集里用“0”表示)
-
第四列母本ID(如果母本不在数据集里用“0”表示)
-
第五列表示性别代码:(’1’=男性,’2’=女性,’0’=未知,也可以用其他数字来凑格式)
-
第六列是表型数据
表型数据
最后还有一个表型的输入文件,一般会用.pheno的后缀表示
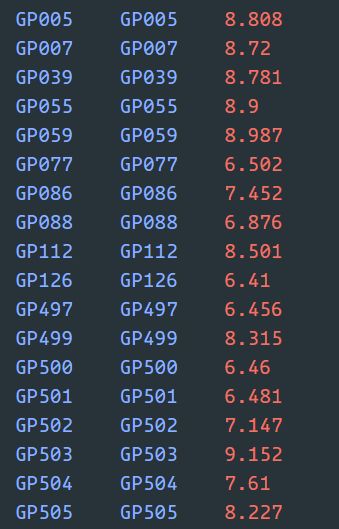
表型文件在每一行有三个条目,FAMID,INDID和表型值。 缺失的表型值应表示为“NA”。(偷懒一点就可以直接用tfam文件里的地1、2、6列)
http://blog.sina.com.cn/s/blog_12d5e3d3c0101qv1u.html





