导读
微生物是人体的重要组成部分,人体微生物的基因数目远远超过人体自身基因数。其中,肠道微生物是人体微生物组的主体,口腔、皮肤、阴道微生物均为人体微生物组的重要组成部分。有研究表明,人体不同部位的微生物组成更是存在很大差异
1
。
人类基因组计划完成之后,人体微生物组研究逐渐提上日程:美国国立卫生研究院于2007年底启动
人体微生物组计划
(
Human Microbiome Project, HMP)旨在通过对人体微生物遗传和代谢的整体研究,了解人体微生物组;紧随其后,欧盟于2008年初启动
人体肠道宏基因组学计划
(Metagenomics of the Human Intestinal Tract, MetaHIT),旨在通过对比疾病人群,解析人体微生物与健康或疾病的关系,促进人类健康。
由于微生物在代谢及免疫调节等方面的重要作用,与人体健康密切相关。所以,近年来,微生物研究也已成为各大科研机构的研究热点,微生物相关产品和应用更是层出不穷。以微生物为研究对象的宏基因组学研究也变得火热起来。
加上,近年来,高通量测序技术的快速发展,使得测序成本大大降低,大规模宏基因组研究更是得以迅速开展。作为宏基因组研究的重要工具,
宏基因组鸟枪法测序技术
使我们能够从基因水平、物种水平、功能水平全方位刻画微生物组;
宏基因组关联分析
作为宏基因组研究的一种重要分析方法,其探究微生物与多种复杂疾病如2型糖尿病、肥胖等的关系,也为疾病预防、诊断、治疗提供了新的思路。
本次特别邀请了深圳华大生命科学研究院宏基因组研究所的研究团队,结合宏基因组已有研究基础与当前宏基因组关联分析的进展,对大规模宏基因组研究思路作一总结和概述。
16s rRNA基因扩增子测序与宏基因组鸟枪法测序
不同于单一物种的基因组研究,宏基因组研究以环境样品中全部微生物基因组为研究对象,其丰富的物种多样性成为宏基因组研究的难点。
16S rRNA基因扩增子测序方法
以分类学标记基因为基础,能够
鉴定样品中存在的微生物种类,研究微生物与疾病之间的关系。
其中,16S rRNA基因扩增子测序相关的研究表明,
肠道菌群失调可能是许多疾病的关键因素
。
然而,该方法产生的数据在低层次的物种分类水平上缺乏一定的分辨能力,加上其产生的数据缺乏功能水平的信息,此方法应用范围有限。
而随着2代测序技术的发展,
宏基因组鸟枪法测序技术
能够
对微生物群落中全部DNA序列进行描述
,
提供所有物种分类水平和功能通路上的基因丰度的信息
,为宏基因组学相关研究的开展提供了技术支持。
宏基因组研究中以基因谱、物种谱和功能谱来描述微生物组,下游分析均以此为基础展开。
如果想让不同样品的基因丰度具有可比性,一个统一且完整的参考基因集显得尤为重要。
2010年,以 MetaHIT 计划为背景,覃俊杰
2
等人
建立了第一个人体肠道菌群非冗余参考基因集
:从124个欧洲人肠道菌群中鉴定到3.3M个微生物基因,是人类基因集的300倍。
【非冗余基因集去除了不同菌种之间的冗余基因以及不同样品之间共有的冗余基因。】该基因集
包含了该人群队列中绝大多数的肠道微生物基因,并且大部分基因在人群中共有
;此外,该研究证实了在宏基因组研究中,短序列可以用于复杂环境中基因的鉴定;并通过对肠道宏基因组和肠细菌基因组进行功能分析,为宏基因组研究确定了基本的研究思路。
在此之后,大多数人类肠道微生物研究都基于参考基因集数据库进行。
但由于数据库构建方法以及样本来源的地域差异,不同的横向研究结果难以比较。2014年,李俊桦
3
等人根据来自三个大洲、共1267个人体肠道微生物样本,结合511个肠道相关的原核生物基因组信息,
构建了一个包含约9.9M个基因的高质量人类肠道微生物基因集数据库(Integrated genecatalog, IGC)
(图1)。
该数据库包含了绝大多数肠道微生物的基因。
以此数据库为基础,该团队发现中国和丹麦人群样本的肠道菌群在物种组成和功能组成上均存在显著差异,
表明地域差异可能造成肠道菌群特征的差异。
2016年,谢海亮
4
等人
对已有的9.9M IGC基因集进行了进一步更新。
他们通过对250名英国成年双胞胎进行粪便菌群宏基因组测序,鉴定到约5.9M个非冗余基因,与9.9M IGC 基因集整合后建立了综合性的肠道菌群参考基因集,发现约11.4M个基因。
非冗余参考基因集的构建和完善为大规模宏基因组研究的开展奠定了基础。
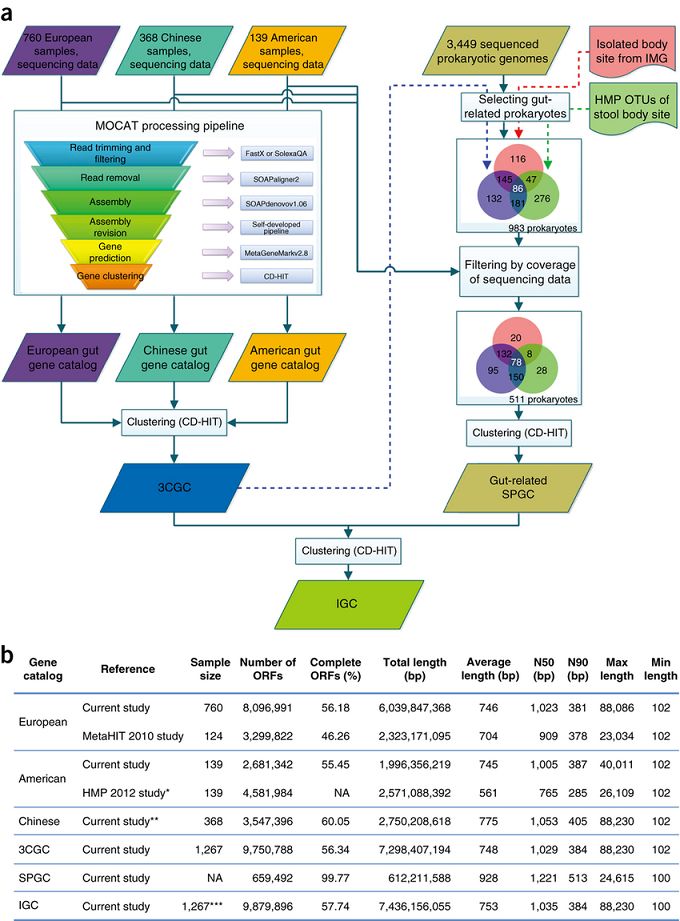
图1 9.9M非冗余参考基因集构建流程
得益于高通量测序技术的快速发展,超大规模的宏基因组研究成为必然趋势,而更大样本量的宏基因组鸟枪法测序依赖于高通量、高性价比的测序平台。
2015年,华大发布新一代测序系统 BGISEQ-500,该测序系统采用了优化的联合探针锚定聚合技术(cPAS)和改进的DNA纳米球(DNB)核心测序技术,新平台在全基因组测序、RNA-seq 及 small RNA-seq 等方面展开全面应用,相关成果陆续在 Cell,GigaScience 等高影响因子杂志刊发表
5,
6,
7
。那么 BGISEQ-500 在宏基因组研究中表现如何?
2017年,方超
15
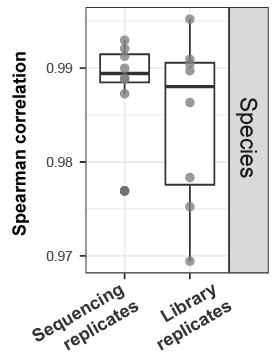
等人对新型高通量测序平台 BGISEQ-500 应用于宏基因组领域的性能进行了综合评估,并将其性能与IlluminaHiSeq 2000与HiSeq 4000平台的性能进行对比。从数据质量、基因丰度、物种丰度等方面分别对平台内稳定性及不同平台的一致性做了评估。结果显示
BGISEQ-500平台内具有极高的技术可重复性
,平台内的建库重复与技术重复的物种丰度相关性高达0.97(图2);跨平台间物种丰度相关性也可达到0.948 (图3),
高准确度及高度技术可重复性表明 BGISEQ500 测序平台对于开展宏基因组研究具有可行性。