转自:Best Media 以及医药领袖

百世传媒(Best Media)在举办仿制药的研发及其质量一致性评价的培训中,培训人员与主讲嘉宾(马小波博士)互动反响强烈。我们会将互动问答内容依次整理到“医药领袖”微信平台,欢迎大家点击右上方关注交流!同时我们会在微信下方发布近期将会举办的培训,想了解具体培训信息的朋友可以点击最下方阅读全文!
马博士,您好!听了您的讲座受益良多,有几个问题向您请教:
1、在日本12年公布的《含量不同的口服固体制剂生物等效性试验指导原则》中提到 “附件2溶出曲线校正方法:当药物溶出有滞后现象时,通常将溶出率达标示量的5%时所需的时间称为“延迟时间”。延迟时间可根据各自的溶出数据、采用内差法求得。当参比制剂与试验制剂的溶出有滞后现象时,应扣除参比制剂与试验制剂每条溶出曲线的延迟时间,求得各自的平均溶出曲线,再进行溶出曲线相似性的比较”,您对这个“延迟时间”怎么解读,在一致性评价过程中能否使用?
答:这是日本12年公布的《含量不同的口服固体制剂生物等效性试验指导原则》中提出的当溶出有滞后现象时,采用延迟时间对溶出曲线予以校正的方法。虽然其他类似指导原则中没有看到有关说明,个人认为是合理的,在一致性评价过程中遇到类似情况可以通过详细说明予以使用。
2、如何进行BCS分类的查询
答: 可以查阅文献。一些文章列出了大量药物的BCS分类资料。下面是一个例子。

3、一个药品在PH1.2中15分钟85%以上,pH4.5 30分钟-60分钟内达到90%,在水中和pH6.8中溶出很低且RSD高,这种情况是这样算是一个比较的结果呢,还是需要改变溶出介质(添加SDS)或选取其他介质进行研究。
答:可以试试改变溶出介质(添加SDS)或选取其他介质进行研究。如果结果不理想,则可能与配方中某种受pH值影响大的辅料含量有关。需要改进配方。
4、一个软胶囊品种,原研欧洲和亚洲上市,FDA网站上有公布其溶出度测定的方法,我们买到三批不同批号的原研产品,参照FDA的方法做发现两个问题:
1) 原研制剂三批差异较大;
2)批内12片溶出10min、15min、20min甚至30min的RSD过大的现象。考虑到剂型的特殊性,我们参照FDA公布的其他软胶囊的溶出方法,我们将取样时间调整为20,30,45,60和90min,并采用了多种方式优化溶出度测定方法如改变测定方法、提高转速、加入胰酶,甚至提高表面活性剂(LDAO)的用量,优化溶出度测定方法,均难以降低RSD,难以采用相似因子f2比较参比制剂和自制制剂溶出曲线的相似性。对于这种批内溶出差异高的制剂老师有什么建议?
答:溶出结果的RSD确实在溶出度试验中很重要。有二大因素将导致溶出结果的变化。一个是制造工艺,另一个是溶出过程。实际上,很难确定哪一个因素起的作用大。可以从溶出曲线对产品制造工艺和溶出方法进行评价。例如,如果早期溶出结果RSD较大,说明方法因素影响较大,如介质,搅拌速度的影响等。如果软明胶胶囊2小时后溶出结果RSD大于20%,3小时后溶出结果RSD 降为0.5%,说明胶囊内颗粒的溶出是主要因素。在胶囊壳开口后,胶囊内颗粒溶出变化较大。而3小时后溶解速度减小,药物释放降低。因此,胶囊内颗粒溶出对RSD影响减小。你的情况可能还是要从方法上下功夫。 象介质,搅拌速度,取样,过滤等都会影响测定结果。
5、在《普通口服固体制剂溶出曲线测定与比较指导原则》其他项下提到“当溶出曲线不能采用相似因子(f2)法比较时,可采用其他适宜的比较法,但在使用时应给予充分论证。”,而《普通口服固体制剂溶出度试验技术指导原则》中也提到的溶出批内差异较大时可采用非模型依赖多变量置信区间法进行溶出比较,想向您请教下这种方法的可行性?能否举例说明如何具体操作?
答:“普通口服固体制剂溶出度试验技术指导原则”中有关非模型依赖多变量置信区间法 对于批内溶出量相对标准偏差大于15%的产品, 可能更适于采用非模型依赖多变量置信区间方法进行溶出曲线比较。建议按照下列步骤进行:
(1)测定参比样品溶出量的批间差异,然后以此为依据确定多变量统计矩(Multivariate statistical distance,MSD)的相似性限度。
(2)确定受试和参比样品平均溶出量的多变量统计矩。
(3)确定受试和参比样品实测溶出量多变量统计矩的90%置信区间。
(4)如果受试样品的置信区间上限小于或等于参比样品的相似性限度,可认为两个批次的样品具有相似性。
比较复杂。找到一篇文章。你自己去读。

Model Independent Approaches One such model is the independent multivariate confidence region procedure (Bootstrap approach) method. Bootstrap allows for the use of f2, not as a point estimator but as a confidence interval, thus overcoming concerns encountered when f2 is used solely as a point estimate. Computation of the bias corrected and accelerated (BCA) confidence intervals is suggested. The bootstrap method simulates the distribution of f2 values to determine whether or not two profiles exhibit comparable in vitro behavior. (6,9) This can include the following steps:
1. Generate N bootstrap samples (e.g., N = 1000) by resampling independently with replacement from dissolution data for the test and reference groups.
2. Calculate f2 values from N bootstrap samples.
3. Calculate the bias correction statistic to correct for the potential skewed distribution of f2 derived from the bootstrap samples. This can be achieved by calculating the acceleration statistic (i.e., the rate of change of the standard error of the estimate of the sample statistic) by obtaining n jackknife samples (e.g., n = 24) derived from original dissolution data
4. Calculate lower and upper bounds of the confidence interval using type 1 error, f2 values of N bootstrap samples, bias correction, and acceleration statistics.
To declare similarity between test and reference dissolution profiles, the lower bound of the confidence interval must equal 50 or more. To illustrate, the bootstrap method was applied by the FDA when they evaluated the reformulated 400 mg mesalamine delayed-release (MDR) (DelzicolTM) against the original 400 mg mesalamine modified-release tablets (10). The newly marketed MDR capsule and discontinued tablet (Asacol®) formulations have a pH-dependent release mechanism designed to delay release until the formulation reaches the distal gastrointestinal (GI) tract (pH~6.5), thus, increasing the level of exposure to the colon in patients with ulcerative colitis. The dissolution characteristics and pharmacokinetics of these MDR capsules and tablets are highly variable. As part of the reformulation program to market the capsule formulation, the similarity of dissolution profiles to the original MDR tablet formulation were needed across various pH values to provide the necessary assurance of comparable product performance (10). For modified-release oral dosage forms such as DelzicolTM that are developed for targeted local drug delivery within the GI tract, the dissolution profiles must contain at least three timepoints (11). As stated in the SUPAC MR 1997 guidance, “Adequate sampling should be performed, for example, at 1, 2, and 4 h and every two hours thereafter until either 80% of the drug from the drug product is released or an asymptote is reached.” (12) (Fig. 1).
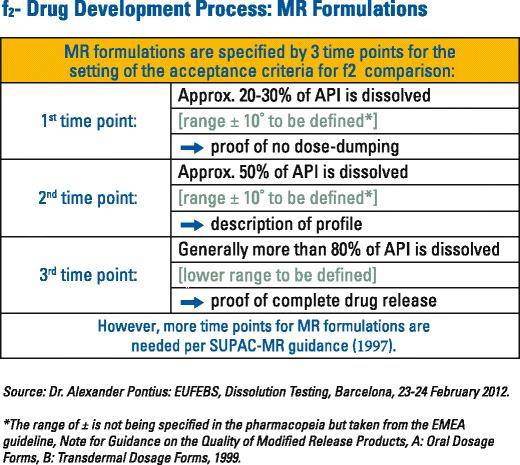
Fig. 1 Modified-release dissolution time points A multipoint dissolution profile comparison was generated between the mesalamine capsules and tablets over a range of pH values (pH 4.5, 6.0, 6.5, 6.8, 7.2, and 7.5). The similarity factors (f2) values were calculated and provided for each dissolution media. Figure 2 displays the dissolution profiles before and after the formulation change at pH at 7.2, for which the f2 value was calculated to be 55.5 by the applicant. The variability (%RSD) in the dissolution data was very high at the 15- (170.1%), 30- (80.9%) and 45- (33.4%) min time points. Because of this, the f2 test could not be used and the FDA applied the model independent bootstrap approach.
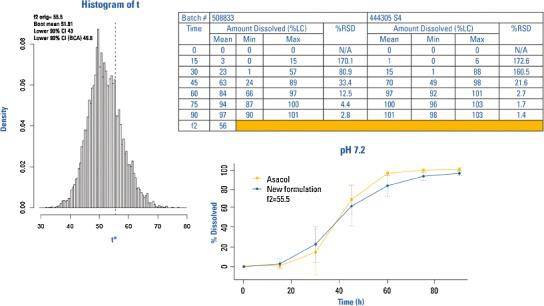
Example: bootstrap versus original method for f2 determination results
Fig. 2 Example: bootstrap versus original method for f2 determination results The bootstrap lower confidence interval with a bias correction was equal to 49.8. The FDA accepted this value in demonstrating similarity of the two products. The bootstrap method should not be used to improve an f2 but rather it should be used as a mechanism for dealing with the issue of data variability. It is noted that there is an open software available for the bootstrap calculation (13).










