N-三氟甲基亚砜邻苯二甲酰亚胺——一种通用的三氟甲基亚砜基化试剂
CF3S(O)n(n = 0, 1, 2)是药物设计中有价值的重要片段,尤其是在将这些部分引入到生物活性化合物中,这主要是因为它们具有很强的吸电子性和特殊的亲脂性。例如,SCF3基团具有高亲脂性(π = 1.44),而SO2CF3表现出强吸电子性(σp = 0.93)。与SCF3和SO2CF3取代基相比,三氟甲基亚砜基(CF3S(O))具有中等的吸电子性和负的亲脂性(π = -0.24),从而使药物化学家能够在新药研发中微调先导化合物的功能性氟烷基的脂溶性。此外,就Hammett取代基常数而言,CF3S(O)的取代基常数(σm = 0.63,σp = 0.69)远高于相近的氟烷基CF3S和CF3。另外 CF3S(O)基团具有CF3S和CF3SO2之间的“中间价态”特征,这使其能够转化为其他重要的氟烷基基团,包括CF3S、CF3SO2和CF3SO(NH)。因此,这些特殊的物理和化学性质的变化是生物活性增强的原因,这可以在各种含有CF3S(O)的农用化学品和药物中找到,例如世界著名且广泛使用的杀虫剂氟虫腈及其其他生物活性分子(图1)。因此,将三氟甲基亚砜基引入所需目标分子已成为药物化学家提高新药发现临床成功前景的不可或缺的策略。图1:含有CF3S(O)基团的生物活性化合物(来源:Org. Chem. Front.)传统上,构建三氟甲基亚砜基化合物的常用策略是:在氟负离子的活化下,利用TMSCF3对亚磺酰基卤化物或亚磺酸酯的亲核三氟甲基化反应。此外,三氟甲基硫化物通过不同氧化体系的氧化策略也可以提供CF3S(O)的产物,但是该过程需要多步骤来合成RSCF3,并且可能伴随不需要的过度氧化的砜(CF3SO2)和其他硫化物的产生。此外,近些年来,来自CF3SO2源的试剂(CF3SO2Na、CF3SO2Cl、BT-SO2CF3,图2,试剂1-3)在磷或硫添加剂如PCl3、TfOH、POCl3、PCy3、Ph2P(O)Cl的存在下,可与多种亲核试剂发生脱氧的三氟甲基亚砜基化反应。然而,这些CF3SO2衍生的试剂没有摆脱潜在过度还原为三氟甲硫基化产物(RSCF3)所特有的缺点,并且其具有低操作温度(试剂2)和多步合成步骤(试剂3)等问题。与此形成鲜明对比的是,在温和的条件下通过使用CF3S(O)试剂直接引入所需的CF3S(O)基团是特别有利的,特别是在含有对氧化剂或脱氧试剂敏感的官能团的复杂反应体系中。尽管已经报道了使用CF3S(O)Cl(试剂4)获得三氟甲基亚砜基化化合物的直接方法,但由于CF3S(O)Cl的生理毒性、高挥发性(31℃)和较差的稳定性,这些特定的困难和局限性也限制了其进一步利用。近些年来也出现了一些直接亲电的三氟甲基亚砜基化试剂来取代这种有问题的CF3S(O)Cl。Larock小组使用基于苯胺的三氟甲基亚磺酰胺作为CF3S(O)源,可以作为双官能团试剂,与苯炔底物顺利生成邻三氟甲基亚砜苯胺化合物。值得注意的是,2003年,Bertrand发现基于丁二酰亚胺的三氟甲基亚砜基化试剂(图2,试剂5),作为CF3S(O)Cl的有价值替代品出现,可以将三氟甲基亚砜基引入到为数不多的有机分子中,如吡咯、醇(由化学计量的路易斯碱Et3N活化),和仲胺(通过化学计量的路易斯碱Me2NH活化)。另外,2023年,杭州师大的邵欣欣课题组通过β-三氟亚磺酰基酯与芳香族或芳基高价碘底物进行了一种新型的亲核三氟甲基亚砜基化反应。然而,直到目前,许多亲核试剂(C、N、O)的直接亲电三氟甲基亚砜基化试剂的选择仍然很少。因此,寻找通用的三氟甲基亚砜基化试剂来有效转移CF3S(O)基团,并探索其活化模式和机理,具有很高的价值和科学意义。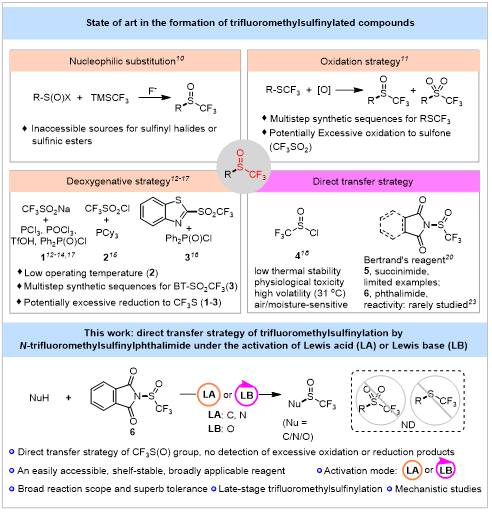 图2:三氟甲基亚砜基化化合物的合成策略(来源:Org. Chem. Front.)N-三氟甲基亚砜邻苯二甲酰亚胺6由Bertrand于2003年合成,但其化学性质和合成应用很少被研究。在此,作者报道了在催化或化学计量的路易斯酸/路易斯碱的活化下,通过N-三氟甲基亚砜邻苯二甲酰亚胺来向多种亲核试剂高效转移三氟甲基亚砜基的策略。N-三氟甲基亚砜邻苯二甲酰亚胺6的优异反应性通过C/N/O亲核试剂的有效三氟甲基亚砜基化进一步证明,所述亲核试剂包括富电子芳杂环、胺、烷基醇和具有挑战性的弱亲核苯酚,与传统试剂相比,没有观察到过度氧化或还原的产物(图2)。作者首先探索了富电子杂芳烃的Friedel-Crafts型三氟甲基亚砜基化反应,因为这代表了一种直接有效的方法,可以将三氟甲基亚砜基引入众多主要候选药物的富电子芳杂环中。作者最初将试剂5或6和吲哚在50°C、无活化剂条件下进行反应,得到了低产率的3-三氟甲基亚砜基化吲哚;然而,当CF3S(O)Cl作为亲电试剂时,以中等产率观察到三氟甲基亚砜基化产物(图3)。此外,作者系统地优化了这种亲电的三氟甲基亚砜基化反应,以在活化剂的帮助下增强亲电性,发现酸性添加剂在这种Friedel-Crafts型三氟甲基亚砜化中是关键的。在路易斯酸如BF3·OEt2、AlCl3、ZnCl2、MgCl2或LiCl以及Brønsted酸(包括TfOH和PTSA)存在下的反应是无效的。令人惊喜的是,在1.5当量Me3SiCl存在下,吲哚与试剂6在50°C下反应16小时后顺利进行,以97%的产率得到3-三氟甲基亚砜化吲哚8aa(图3)。当作者使用Me3SiBr代替Me3SiCl作为活化剂时,没有观察到期望的产物。作为比较,在Me3SiCl的活化下,Bertrand试剂5仅提供82%产率的3-三氟甲基亚砜基化吲哚。此外,溶剂的选择对于Friedel-Crafts型三氟甲基亚砜基化至关重要:烷基氯代溶剂(尤其是CHCl3)有助于该反应,而在甲苯中也可以获得令人满意的产率,并且在极性溶剂如THF中没有观察到产物(图3)。正如预期,这种亲电的三氟甲基亚砜基化可以在室温下进行,尽管产率略有下降。图3: 反应条件的优化(来源:Org. Chem. Front.)在优化的条件下,各种吲哚与3-、4-、5-、6-或7-位给电子或吸电子取代基的反应顺利进行,以良好的定量产率(43-99%)生成了三氟甲基亚砜基化吲哚(8aa-8bf,图4)。该反应显示出优异的官能团相容性,许多官能团如甲氧基、苄氧基、碳酸叔丁酯、硼酸频哪醇、氨基、乙烯基、氟、氯、溴、酯基,甚至是未保护的羟基,都可以兼容,以良好至高产率得到相应的产物(8aa-8bd)。值得注意的是,试剂6与5-(2,6-二甲氧基苯基)-1H-吲哚或1'H-1,5'-双吲哚的反应显示出相对于苯环或缺电子吲哚,反应对富电子的吲哚具有优异的选择性,分别提供45%和53%的产率(8be-8bf)。试剂6与吡咯、1H-吡咯并[2,3-b]-吡啶、咪唑并[1,2-a]-吡啶和异恶唑在内的其他富电子杂芳烃,以72−99%的产率成功地得到了相应的三氟甲基亚砜基化杂环产物(8bg-8bl,图4)。然而,在这些条件下富电子芳烃如1,3-二甲氧基苯或甲氧基苯未能检测到目标的三氟甲基亚砜基化产物(8bm-8bn)。图4:富电子杂芳烃底物范围(来源:Org. Chem. Front.)与发展较为成熟的C-三氟甲基亚砜基化相比,杂原子(N,O)的三氟甲基砜基化的研究则相对较少。随着三氟甲基亚砜基化芳杂环合成方法的成功发展,作者接下来试图研究胺与试剂6的亲电三氟甲基亚砜基化反应。与使用路易斯碱(Me2NH)活化试剂5和仲胺的文献不同,作者发现路易斯酸Ph2P(O)Cl非常适合烷基胺和试剂6的三氟甲基亚砜基化,在筛选反应条件后,可提供85%产率的N-S(O)CF3产物。相比之下,Langlois试剂(CF3SO2Na)作为三氟甲基亚砜基源,与胺的尝试可以得到70%收率的4-碘苯基三氟甲基亚磺酰胺,而CF3S(O)Cl只得到了5%的产率。带有不同卤素、氰基、三氟甲基、甲氧基、叔丁基的富电子和缺电子苄胺与试剂6反应,以38-79%的产率得到相应的三氟甲基亚砜化产物(10aa-10ak,图5)。环烷基胺(1-茚胺、环己胺、4-氨基-1-boc-哌啶)和包括苯乙胺和乙酰胺在内的长链烷基胺的亲电三氟甲基亚砜基化反应都可以顺利发生(10al-10ap)。此外,氨基酯也可以实现三氟甲基亚砜基化反应,以44%的产率(10aq)提供所需的三氟甲基亚磺酰胺。呋喃胺、噻吩-2-乙胺和色胺的杂环胺底物在该体系中也是相容的(10ar-10at)。除了伯烷基胺外,仲烷基胺包括N-甲基-1-苯基甲烷胺、二苄基胺、哌啶和1,2,3,4-四氢异喹啉与6的反应在标准条件下也可以很好地进行反应(10au-10ax)。随后,作者在相同的反应条件下也研究了具有优异官能团耐受性的弱亲核芳胺的底物范围。这些转化以中高产率提供了三氟甲基亚磺酰胺,收率与烷基胺相比略有增加。多种富含电子或缺乏电子的苯胺与试剂6反应,在温和的条件下以良好至优异的产率成功地得到相应的三氟甲基亚砜化产物。具有吸电子取代基的芳胺的反应比具有富电子基团的芳胺要快得多(10ay-10bn)。此外,当作者使用萘-2-胺作为亲核性胺底物(10bo)时,可以获得57%的期望产物的产率。苯并[d]噻唑-5-胺、2-苯并噻唑胺、5-氨基嘧啶和3-氨基喹啉在内的含氮杂芳基胺在标准条件下也可有效转化为三氟甲基亚砜基取代的杂芳胺(10bp-10bs)。仲环芳香胺如吲哚啉和1,2,3,4-四氢喹啉,分别以89%和85%的产率提供N-三氟甲基亚砜化产物(10bt-10bu)。值得注意的是,Larock试剂的类似骨架胺(N-甲基苯胺)也可以以中等产率制备1,1,1-三氟-N-甲基-N-苯基甲亚磺酰胺(10bv)。图5:胺类底物范围(来源:Org. Chem. Front.)在试剂6与芳胺/烷基胺优异反应性能的鼓舞下,作者进一步研究了醇的亲电三氟甲基亚砜基化反应。当使用CF3S(O)Cl时仅得到20%的目标产物产率,并且大量的4-碘苄醇仍未转化。值得注意的是,与试剂5相比,催化量的Et3N可以成功有效地促进试剂6对烷基醇的三氟甲基亚砜化反应。醇与试剂6在10 mol% Et3N存在下,在THF中室温反应3小时可以得到中等至高产率的目标产物,无需路易斯酸活化剂的帮助,如图6所示。不同取代的苄醇以42-99%的产率转化为相应的三氟甲基亚磺酸酯,可以兼容氟、氯、碘、三氟甲基、醛、三氟甲基磺酰基、酯、氰基、硝基、频哪醇硼酸酯、苯氧基和甲氧基(12aa-12ao)。此外,萘-1-基甲醇可以中等的产率成功得到相应的产物(12ap)。类似地,长链烷基醇如苯乙醇和苯丙醇在该体系中也是有效的(12aq-12at),表明试剂6具有极好的烷基醇底物范围。此外,杂环烷基醇如2-(噻吩-3-基)乙烷-1-醇和色氨醇也能顺利发生三氟甲基亚砜化反应,提供80-81%产率的三氟甲基亚磺酸酯(12au-12av)。对于具有远端烯基的香茅醇,也可生成高产率的三氟甲基亚磺酸酯(12aw)。除了伯醇之外,仲醇(DL-扁桃酸甲酯、9-芴醇、2-茚醇)和叔醇(1-金刚烷醇)可以在温和的反应条件下方便地进行三氟甲基亚砜化反应(12ax-12az,12bc)。此外,简单的天然仲醇L-薄荷脑和冰片与试剂6以高至优异的产率进行反应(84%,12ba;77%,12bb)。值得注意的是,在该体系中,没有观察到三氟甲基亚磺酸烷基酯的任何分解或重排,可以通过三氟甲基亚磺酸苄酯(12ae)的单晶X射线结构得到了明确的证实。对于更具挑战性的弱亲核性苯酚底物,通过使用的CF3S(O)试剂来获得三氟甲基亚磺酸苯酯化合物的直接方法很少被报道。有趣的是,当作者将O-三氟甲基亚砜化扩展到酚类底物时,发现富电子基取代的酚类可以在50 mol%Et3N和加热条件(50℃)下,通过这种稳定的N-三氟甲基亚砜邻苯二甲酰亚胺顺利产生所需的亲电三氟甲基亚砜化产物。多种官能团如甲氧基、甲硫基、苄氧基、苯氧基、异丙基、叔丁基、苄基、环烷基甚至轻微吸电子基团(苯基和碘)可以兼容该体系,以中等至优异的产率获得目标产物(12bd-12bq)。然而,缺电子的苯酚不能发生这种亲电的三氟甲基亚砜化反应。需要注意的是,分离的三氟甲基亚磺酸苯酯相对不稳定,需要保存在冰箱中。图6:烷基醇以及酚类底物范围(来源:Org. Chem. Front.)该策略具有良好的官能团耐受性和高效性,作者也成功将其应用于天然产物或生物活性分子的后期三氟甲基亚砜基化反应中(图7)。
图2:三氟甲基亚砜基化化合物的合成策略(来源:Org. Chem. Front.)N-三氟甲基亚砜邻苯二甲酰亚胺6由Bertrand于2003年合成,但其化学性质和合成应用很少被研究。在此,作者报道了在催化或化学计量的路易斯酸/路易斯碱的活化下,通过N-三氟甲基亚砜邻苯二甲酰亚胺来向多种亲核试剂高效转移三氟甲基亚砜基的策略。N-三氟甲基亚砜邻苯二甲酰亚胺6的优异反应性通过C/N/O亲核试剂的有效三氟甲基亚砜基化进一步证明,所述亲核试剂包括富电子芳杂环、胺、烷基醇和具有挑战性的弱亲核苯酚,与传统试剂相比,没有观察到过度氧化或还原的产物(图2)。作者首先探索了富电子杂芳烃的Friedel-Crafts型三氟甲基亚砜基化反应,因为这代表了一种直接有效的方法,可以将三氟甲基亚砜基引入众多主要候选药物的富电子芳杂环中。作者最初将试剂5或6和吲哚在50°C、无活化剂条件下进行反应,得到了低产率的3-三氟甲基亚砜基化吲哚;然而,当CF3S(O)Cl作为亲电试剂时,以中等产率观察到三氟甲基亚砜基化产物(图3)。此外,作者系统地优化了这种亲电的三氟甲基亚砜基化反应,以在活化剂的帮助下增强亲电性,发现酸性添加剂在这种Friedel-Crafts型三氟甲基亚砜化中是关键的。在路易斯酸如BF3·OEt2、AlCl3、ZnCl2、MgCl2或LiCl以及Brønsted酸(包括TfOH和PTSA)存在下的反应是无效的。令人惊喜的是,在1.5当量Me3SiCl存在下,吲哚与试剂6在50°C下反应16小时后顺利进行,以97%的产率得到3-三氟甲基亚砜化吲哚8aa(图3)。当作者使用Me3SiBr代替Me3SiCl作为活化剂时,没有观察到期望的产物。作为比较,在Me3SiCl的活化下,Bertrand试剂5仅提供82%产率的3-三氟甲基亚砜基化吲哚。此外,溶剂的选择对于Friedel-Crafts型三氟甲基亚砜基化至关重要:烷基氯代溶剂(尤其是CHCl3)有助于该反应,而在甲苯中也可以获得令人满意的产率,并且在极性溶剂如THF中没有观察到产物(图3)。正如预期,这种亲电的三氟甲基亚砜基化可以在室温下进行,尽管产率略有下降。图3: 反应条件的优化(来源:Org. Chem. Front.)在优化的条件下,各种吲哚与3-、4-、5-、6-或7-位给电子或吸电子取代基的反应顺利进行,以良好的定量产率(43-99%)生成了三氟甲基亚砜基化吲哚(8aa-8bf,图4)。该反应显示出优异的官能团相容性,许多官能团如甲氧基、苄氧基、碳酸叔丁酯、硼酸频哪醇、氨基、乙烯基、氟、氯、溴、酯基,甚至是未保护的羟基,都可以兼容,以良好至高产率得到相应的产物(8aa-8bd)。值得注意的是,试剂6与5-(2,6-二甲氧基苯基)-1H-吲哚或1'H-1,5'-双吲哚的反应显示出相对于苯环或缺电子吲哚,反应对富电子的吲哚具有优异的选择性,分别提供45%和53%的产率(8be-8bf)。试剂6与吡咯、1H-吡咯并[2,3-b]-吡啶、咪唑并[1,2-a]-吡啶和异恶唑在内的其他富电子杂芳烃,以72−99%的产率成功地得到了相应的三氟甲基亚砜基化杂环产物(8bg-8bl,图4)。然而,在这些条件下富电子芳烃如1,3-二甲氧基苯或甲氧基苯未能检测到目标的三氟甲基亚砜基化产物(8bm-8bn)。图4:富电子杂芳烃底物范围(来源:Org. Chem. Front.)与发展较为成熟的C-三氟甲基亚砜基化相比,杂原子(N,O)的三氟甲基砜基化的研究则相对较少。随着三氟甲基亚砜基化芳杂环合成方法的成功发展,作者接下来试图研究胺与试剂6的亲电三氟甲基亚砜基化反应。与使用路易斯碱(Me2NH)活化试剂5和仲胺的文献不同,作者发现路易斯酸Ph2P(O)Cl非常适合烷基胺和试剂6的三氟甲基亚砜基化,在筛选反应条件后,可提供85%产率的N-S(O)CF3产物。相比之下,Langlois试剂(CF3SO2Na)作为三氟甲基亚砜基源,与胺的尝试可以得到70%收率的4-碘苯基三氟甲基亚磺酰胺,而CF3S(O)Cl只得到了5%的产率。带有不同卤素、氰基、三氟甲基、甲氧基、叔丁基的富电子和缺电子苄胺与试剂6反应,以38-79%的产率得到相应的三氟甲基亚砜化产物(10aa-10ak,图5)。环烷基胺(1-茚胺、环己胺、4-氨基-1-boc-哌啶)和包括苯乙胺和乙酰胺在内的长链烷基胺的亲电三氟甲基亚砜基化反应都可以顺利发生(10al-10ap)。此外,氨基酯也可以实现三氟甲基亚砜基化反应,以44%的产率(10aq)提供所需的三氟甲基亚磺酰胺。呋喃胺、噻吩-2-乙胺和色胺的杂环胺底物在该体系中也是相容的(10ar-10at)。除了伯烷基胺外,仲烷基胺包括N-甲基-1-苯基甲烷胺、二苄基胺、哌啶和1,2,3,4-四氢异喹啉与6的反应在标准条件下也可以很好地进行反应(10au-10ax)。随后,作者在相同的反应条件下也研究了具有优异官能团耐受性的弱亲核芳胺的底物范围。这些转化以中高产率提供了三氟甲基亚磺酰胺,收率与烷基胺相比略有增加。多种富含电子或缺乏电子的苯胺与试剂6反应,在温和的条件下以良好至优异的产率成功地得到相应的三氟甲基亚砜化产物。具有吸电子取代基的芳胺的反应比具有富电子基团的芳胺要快得多(10ay-10bn)。此外,当作者使用萘-2-胺作为亲核性胺底物(10bo)时,可以获得57%的期望产物的产率。苯并[d]噻唑-5-胺、2-苯并噻唑胺、5-氨基嘧啶和3-氨基喹啉在内的含氮杂芳基胺在标准条件下也可有效转化为三氟甲基亚砜基取代的杂芳胺(10bp-10bs)。仲环芳香胺如吲哚啉和1,2,3,4-四氢喹啉,分别以89%和85%的产率提供N-三氟甲基亚砜化产物(10bt-10bu)。值得注意的是,Larock试剂的类似骨架胺(N-甲基苯胺)也可以以中等产率制备1,1,1-三氟-N-甲基-N-苯基甲亚磺酰胺(10bv)。图5:胺类底物范围(来源:Org. Chem. Front.)在试剂6与芳胺/烷基胺优异反应性能的鼓舞下,作者进一步研究了醇的亲电三氟甲基亚砜基化反应。当使用CF3S(O)Cl时仅得到20%的目标产物产率,并且大量的4-碘苄醇仍未转化。值得注意的是,与试剂5相比,催化量的Et3N可以成功有效地促进试剂6对烷基醇的三氟甲基亚砜化反应。醇与试剂6在10 mol% Et3N存在下,在THF中室温反应3小时可以得到中等至高产率的目标产物,无需路易斯酸活化剂的帮助,如图6所示。不同取代的苄醇以42-99%的产率转化为相应的三氟甲基亚磺酸酯,可以兼容氟、氯、碘、三氟甲基、醛、三氟甲基磺酰基、酯、氰基、硝基、频哪醇硼酸酯、苯氧基和甲氧基(12aa-12ao)。此外,萘-1-基甲醇可以中等的产率成功得到相应的产物(12ap)。类似地,长链烷基醇如苯乙醇和苯丙醇在该体系中也是有效的(12aq-12at),表明试剂6具有极好的烷基醇底物范围。此外,杂环烷基醇如2-(噻吩-3-基)乙烷-1-醇和色氨醇也能顺利发生三氟甲基亚砜化反应,提供80-81%产率的三氟甲基亚磺酸酯(12au-12av)。对于具有远端烯基的香茅醇,也可生成高产率的三氟甲基亚磺酸酯(12aw)。除了伯醇之外,仲醇(DL-扁桃酸甲酯、9-芴醇、2-茚醇)和叔醇(1-金刚烷醇)可以在温和的反应条件下方便地进行三氟甲基亚砜化反应(12ax-12az,12bc)。此外,简单的天然仲醇L-薄荷脑和冰片与试剂6以高至优异的产率进行反应(84%,12ba;77%,12bb)。值得注意的是,在该体系中,没有观察到三氟甲基亚磺酸烷基酯的任何分解或重排,可以通过三氟甲基亚磺酸苄酯(12ae)的单晶X射线结构得到了明确的证实。对于更具挑战性的弱亲核性苯酚底物,通过使用的CF3S(O)试剂来获得三氟甲基亚磺酸苯酯化合物的直接方法很少被报道。有趣的是,当作者将O-三氟甲基亚砜化扩展到酚类底物时,发现富电子基取代的酚类可以在50 mol%Et3N和加热条件(50℃)下,通过这种稳定的N-三氟甲基亚砜邻苯二甲酰亚胺顺利产生所需的亲电三氟甲基亚砜化产物。多种官能团如甲氧基、甲硫基、苄氧基、苯氧基、异丙基、叔丁基、苄基、环烷基甚至轻微吸电子基团(苯基和碘)可以兼容该体系,以中等至优异的产率获得目标产物(12bd-12bq)。然而,缺电子的苯酚不能发生这种亲电的三氟甲基亚砜化反应。需要注意的是,分离的三氟甲基亚磺酸苯酯相对不稳定,需要保存在冰箱中。图6:烷基醇以及酚类底物范围(来源:Org. Chem. Front.)该策略具有良好的官能团耐受性和高效性,作者也成功将其应用于天然产物或生物活性分子的后期三氟甲基亚砜基化反应中(图7)。图7:天然产物或生物活性分子的后期三氟甲基亚砜基化反应(来源:Org. Chem. Front.)
为了阐明路易斯酸/路易斯碱催化或介导的C/N/O亲核试剂的三氟甲基亚砜化的反应机理,作者随后进行了19F NMR光谱跟踪实验。通过19F NMR实验(图8 A-B),作者观察到市售的CF3S(O)Cl在−77.0ppm处,而试剂6的峰在-69.2 ppm处。当将Me3SiCl添加到试剂6和CHCl3的溶液中时,50℃反应4 h后在−77.0 ppm处出现尖锐的单峰(图8 C),这与19F NMR光谱观察到的CF3S(O)Cl的峰位置高度一致。此外,将反应时间延长至10 h时,−77.0 ppm的可能活性中间体缓慢增加(图8 D)。随后,在向图8 D的混合物中加入吲哚底物后,在50℃下继续反应6 h。试剂6的峰消失,−77.0ppm处的单峰相应减少,而所得三氟甲基亚砜基化吲哚(-73.5ppm)的新峰以高产率出现(图8 E),这些说明−77.0pm处的活性物质很可能是CF3S(O)Cl。同样,对于胺的三氟甲基亚砜化反应,通过19F NMR实验(图9 A-B)检测到,THF中的CF3S(O)Cl在−77.8ppm处,而试剂6的峰在-70.7ppm处。有趣的是,当试剂6和Ph2P(O)Cl在室温下反应4-10 h时,CF3S(O)Cl在−77.8 ppm时作为活性中间体出现。在加入4-碘苯胺后,试剂6被快速消耗,并以高产率产生相应的三氟甲基亚磺酰胺(-73.5 ppm),同时在−77.8ppm的信号降低(图9 E),这些也证明了胺的亲电三氟甲基亚砜基化在−77.8ppm的活性物质很可能是CF3S(O)Cl。图8:吲哚的三氟甲基亚砜化反应的19F NMR跟踪实验(来源:Org. Chem. Front.)
此外,通过19F NMR光谱可以看出,4-碘苄醇和1.0当量的试剂6混合,则不会发生反应(图10 A)。有趣的是,当试剂6和1.0当量的Et3N混合10 min至12 h时,则出现了一个新的峰(通常在−88.9 ppm)(图10 B-C),在加入4-碘苄醇后以约42%的产率迅速形成所需的O-S(O)CF3产物(图10 D)。结合作者之前的工作(Org. Lett., 2023, 25, 1066−1071),这些结果表明,由试剂6和Et3N原位生成的高活性物质——三氟甲基亚砜季胺络合物(−88.9 ppm)很可能是将CF3S(O)基团转移到目标醇的活化中间体。图9:胺的三氟甲基亚砜化反应的19F NMR跟踪实验(来源:Org. Chem. Front.)图10:醇的三氟甲基亚砜化反应的19F NMR跟踪实验(来源:Org. Chem. Front.)根据19F NMR跟踪实验和相关文献,作者提出了富电子芳杂环、胺和醇的Friedel-Crafts型三氟甲基亚砜化的合理反应机理,如图11所示。在该机制中,试剂6与Me3SiCl反应,缓慢形成可能的关键中间体CF3S(O)Cl(通过19F NMR实验检测,在−77.0 ppm下观察到)。然后,CF3S(O)Cl在50℃下与吲哚快速反应,得到所需的三氟甲基亚砜化产物(图11 A)。研究发现,吲哚的加入可以加速该亲电的三氟甲基亚砜化反应。类似地,试剂6和Ph2P(O)Cl混合也可以产生关键中间体CF3S(O)Cl(通过19F NMR实验检测,在−77.8 ppm处观察到),与胺发生亲电三氟甲基亚砜基化反应得到目标的三氟甲基亚磺酰胺(图11B)。关于醇的三氟甲基亚砜化反应,通过19F NMR实验观察到了潜在的关键物种(−88.9 ppm)。作者认为三氟甲基亚砜季铵离子对可能是这种亲电的三氟甲基亚砜基化反应进行的原因(图11C)。图11:可能的反应机理(来源:Org. Chem. Front.)朱佃虎课题组发展了一种广谱通用的亲电型三氟甲基亚砜基化试剂,并对不同底物的三氟甲基亚砜化反应的机理进行了初步研究。该试剂具有合成简单、稳定性好、活性高以及应用广泛等优点,可以高效地将三氟甲基亚砜基直接转移至多种亲核试剂,包括富电子芳杂环、烷基胺、芳胺、烷基醇和弱亲核性的苯酚类底物。此外,复杂的生物活性分子也能顺利进行后期的三氟甲基亚砜基化反应,且具有很好的官能团耐受性,这突出了该试剂对于合成化学家在新药开发方面的巨大潜力。本篇工作通讯作者为西北大学的朱佃虎教授和王庆玲博士。西北大学2020级硕士研究生杨柳青和王铄、2022级硕士研究生梁方灿为论文的共同第一作者。上述工作得到了国家自然科学基金(21901201、32200119)、陕西省科技厅青年项目(2020JQ-571)、陕西省教育厅重点实验室等项目(20JS146、21JK0930)经费的大力支持。我们也衷心地感谢西北大学的栾新军教授和中科院上海有机所的沈其龙研究员对我们工作的大力帮助和支持。Direct Transfer Strategy of Trifluoromethylsulfinyl Group by a General Reagent N-Trifluoromethylsulfinylphthalimide under Catalytic or Stoichiometric Lewis Acid or Lewis Base. Liuqing Yang‡, Shuo Wang‡, Fangcan Liang‡, Ying Han, Yilong Li, Dingjian Shan, Lulu Liu, Qingling Wang* and Dianhu Zhu*Org. Chem. Front. 2023, DOI: 10.1039/D3QO00908D.








