
自由基对简单烯烃的加成反应可以有效增加分子的复杂度,该反应最早可以追溯到1945年,Kharasch教授发展了多卤甲烷对末端烯烃的加成,该反应在构建新的C-C键和C-X键的同时可以引入一个CX
3
官能团。该反应作为最早发现的自由基反应之一,后来被命名为Kharasch加成反应,也称为原子自由基加成反应(Scheme 1)。如何实现不对称的Kharasch加成反应是自由基化学领域具有挑战性的历史难题,
德州大学西南医学中心
的
Joseph Ready
教授课题组和
匹兹堡大学
的
刘鹏
教授课题组合作通过实验和计算手段解决了这一难题。这是首例报道高对映选择性的Kharasch加成反应,并首次揭示了外层(out-sphere)自由基的卤素迁移机理(Scheme 2)。
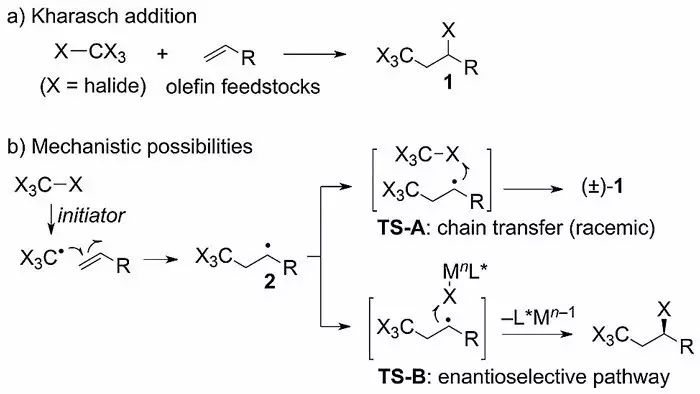
Scheme 1. 多卤甲烷(CX
4
)与烯烃的加成反应。
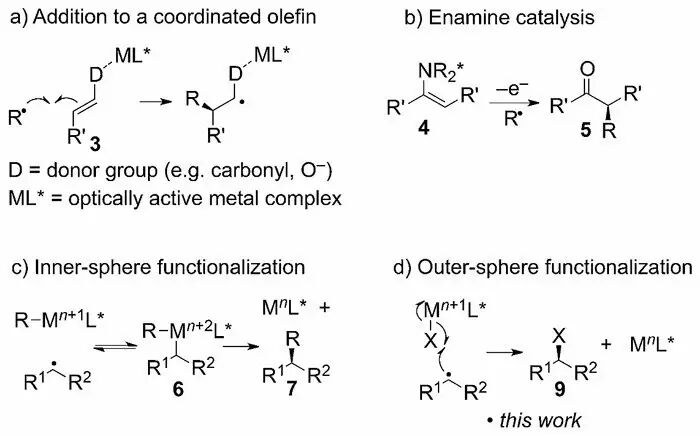
Scheme 2. 不对称自由基反应的机理。a) 自由基与金属结合的底物加成;b)手性烯胺中间体的氧化以及自由基捕获;c)金属捕获自由基中间体及还原消除;d)外层潜手性自由基诱导的原子转移。
首先,他们使用BrCCl
3
与苯乙烯作为起始原料,考察一系列氧化还原过渡金属,如Fe、Mn、Ni、Rh、Ru、Ir和Cu,最终发现Rh具有很高的催化活性,能够在室温以及更低的温度下催化该反应。作者随后在-78 ℃条件下,使用{Rh(cod)Cl}
2
作为催化剂,考察不同单齿及双齿配体的活性,并发现使用单齿膦配体无法参与催化反应。此外BINAP、DuPhos以及Trost配体也没有明显的活性。相反,双齿螯合的双芳基单烷基膦配体Ph
2
P(CH
2
)
n
PPh
2
参与反应具有较好的催化活性,当n=4时,反应的活性最高。作者认为这是由于碳链的长度可以影响配位的螯合键角,从而影响反应的活性。他们以此为基础,使用DIOP作为手性配体,反应产物的er达到89:11。通过进一步的配体优化,作者发现在DIOP苯环的对位引入甲基可以达到最优的效果,位阻小的给电子基团(如甲基和甲氧基)可以提高反应的对映选择性,位阻较大的烷基(如乙基、异丙基和叔丁基)或卤素会降低反应的对映选择性;在DIOP的其他位置进行修饰,如苯环的邻位和间位等都无法进一步提高反应的对映选择性。作者进一步对反应条件进行优化,发现1:1的甲苯与正己烷混合溶剂可以小幅度提高产物的对映选择性,优化其他反应参数如温度、其他Rh催化剂以及不同的溶剂都没有进一步提高反应的选择性(Scheme 3)。
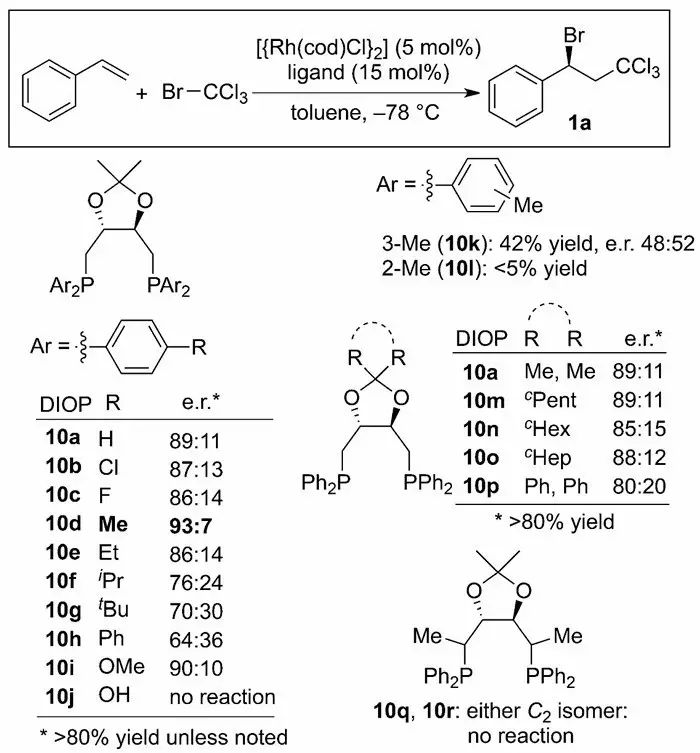
Scheme 3. 对映选择性Kharasch反应的发现。
确定最优条件后,作者进一步考察了反应的底物适用性(Scheme 4a)。该反应可以兼容一系列的官能团,如烷基、卤素、甲氧基以及三甲基硅基(
1b-n
)。芳香环的取代基可以是邻、间、对位取代(
1b-d
),其中对位取代基可以是给电子和中等吸电子基团,强吸电子基团的对映选择性较差。多取代的苯乙烯也可以参与该反应(
1q-s
)。杂环化合物,如3-烯基吡啶可以中等的产率以及对映选择性得到相应的产物(
1t
)。3-烯基吲哚得到的产物(
1u
)不稳定,2-烯基萘反应能以优秀的收率得到目标产物,但1-烯基萘得到的产物(
1p
)不稳定。
其他多卤代烷基也可以引入反应体系中,如-C
Cl
3
、-CBrCl
2
、-CBr
2
Cl、-CBr
3
,以类似的结果得到相应的产物(
1v-x
)。在-78 ℃的标准条件下,苄基溴化物是唯一的产物,并未观察到任何苄基氯化物形成。
苄基溴化物作为立体专一的S
N
2取代的反应底物具有很高的合成价值。作者初步考察该产物在一系列亲核试剂参与反应条件下的活性(Scheme 4b)。C-Br键可以被转化为C-N键以及C-S键(产物
11
、
12
),而尝试使用碳亲核试剂(氰根、烯醇负离子)会导致底物发生消除反应。
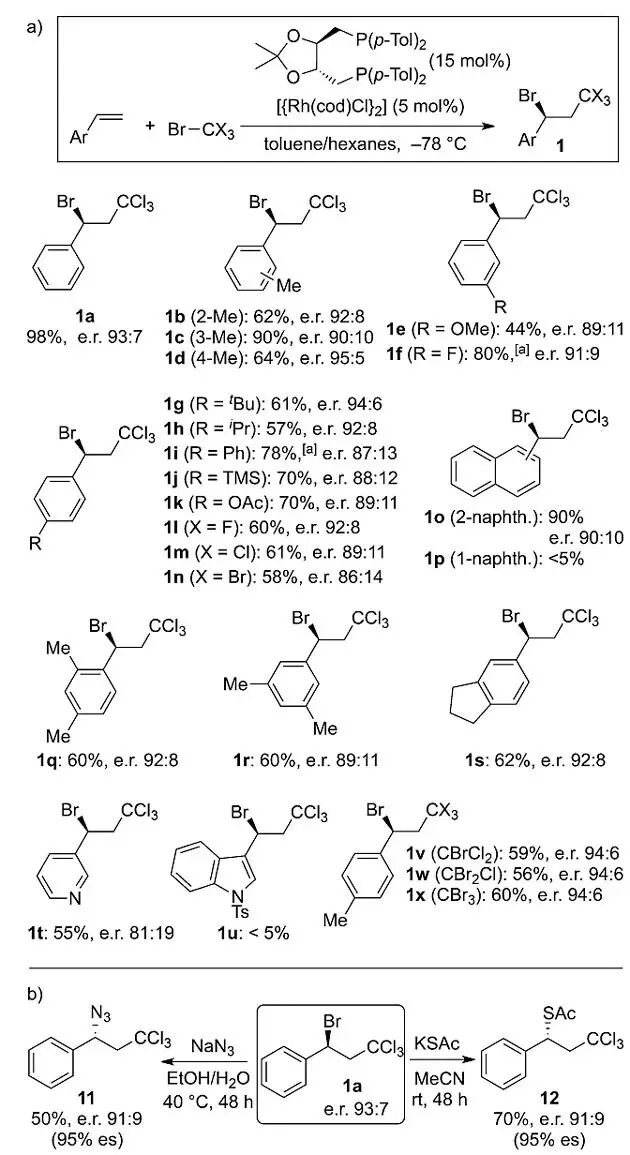
Scheme 4. a)芳基乙烯与CX
4
加成反应的底物普适性考察;b)次级溴化物
1a
的衍生化。
作者推测该反应可能有两种机理,一种是闭壳层的Rh(I)-Rh(III)机理,另一种是自由基机理。第一种机理中首先发生氧化加成生成Br-Rh(III)-C
Cl
3
物种,随后立体专一以及对映选择地插入烯烃得到苄基Rh物种
14
,最后还原消除得到目标产物
1
。第二种机理中首先发生单电子转移产生C
Cl
3
自由基以及Rh(II)-BrCl物种,C
Cl
3
自由基对苯乙烯加成得到更稳定的苄基自由基。此时,苄基自由基可以继续与Rh(II)-BrCl结合生成Rh(III)物种,还原消除得到目标产物
1
,苄基自由基也可能从Rh(II)BrCl物种直接攫取溴原子得到目标产物
1
。
实验数据支持自由基机理,如苯乙烯与CCl
4
及CBr
4
混合,在标准条件下可以得到交叉产物
1a
。在低温条件下,苄基氯化合物并没有产生。作者提出自由基C
Cl
3
对苯乙烯加成得到苄基自由基,该自由基可以从Rh(II)BrCl物种攫取溴原子得到交叉产物
1
,其中C
Cl
3
自由基来自C
Cl
4
,Rh(II)BrCl来自C
Br
4
。反应产率低的主要原因是Rh(I)与C
Cl
4
反应生成的Rh(II)
Cl
2
物种没有活性。作者也进行了同位素标记实验,使用立体构型纯的反式单氘代甲苯与BrC
Cl
3
混合可以得到1:1的非对映异构体。这些实验结果可以排除Rh(I)-Rh(III)机理,因为烯烃插入将会得到立体选择性的产物,而自由基对烯烃加成则会得到无选择性的产物。
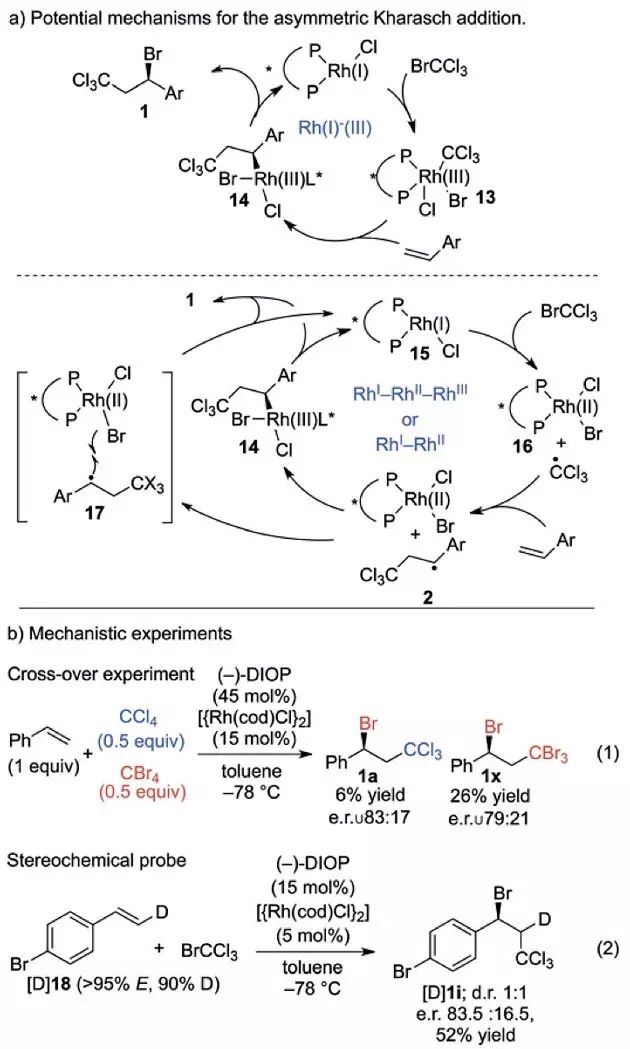
Scheme 5. a)可能的Rh
I
-Rh
III
催化循环机理(上)以及两种自由基中间体参与的催化循环机理(下);b)加成反应机理的探究。
与此同时,刘鹏教授课题组利用DFT计算研究该反应的机理,并以此探索苯乙烯对映选择性溴化的关键步骤(Figure 1)。首先,催化剂[(-)-DIOP]RhCl(
15
)与BrC
Cl
3
作用生成
22
,
22
通过溴原子转移的自由基机理生成Rh(II)-BrCl (
16
)和C
Cl
3
自由基,该过程需要活化能13.8 kcal/mol(
23-TS
)。相比之下,闭壳层的Br-C
Cl
3
发生氧化加成的过渡态(
24-TS
)或者类似
S
N
2
取代反应的直线型过渡态(
25-TS
)都需要更高的能垒,因而反应不能发生。随后,C
Cl
3
自由基对苯乙烯加成生成更稳定的苄基自由基(
2a
),发生开壳层溴原子转移的自由基机理(经历过渡态
17-TS
)与竞争的闭壳层还原消除机理(先生成Rh(III)中间体,再经历过渡态
27-TS
)更具有优势。综上所述,DFT计算揭示了该Kharasch加成反应遵循两步溴原子转移的自由基Rh(I)-Rh(II)催化机理。相比之下,闭壳层的过渡态(
24-TS
、
25-TS
以及
27-TS
)由于Rh中心与C
Cl
3
或者苯环间不利的空间排斥作用较不稳定。
有趣的是,该反应机理中两个溴原子转移的自由基过渡态(
23-TS
和
17-TS
)并非直线型(不同于典型的自由基转移过渡态),Rh-Br-C的角度大约是85°-105°。这种非线型的过渡态结构导致手性双膦配体与底物之间更强的相互作用(
17-TS
),从而对Rh催化剂的手性诱导起到至关重要的作用。在S构型的过渡态(
17-TS-
S
)中,由于存在苄基自由基的苯环与DIOP配体中苯环间的π-π相互作用,使该过渡态能量更低。反之,在R构型的过渡态(
17-TS-
R
)中,苄基自由基则远离配体,缺少稳定的π-π相互作用。
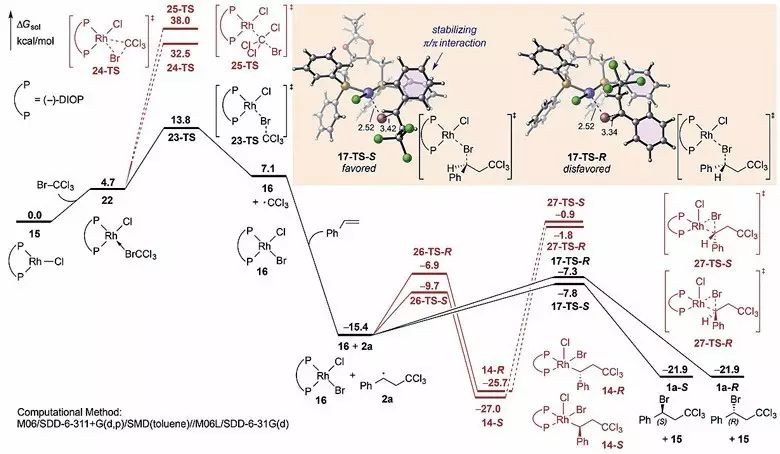
Figure 1. (DIOP)RhCl催化苯乙烯溴化机理的理论计算势能面:自由基机理(黑色),闭壳层机理(红色)。
综上所述,该论文报道了首例高对映选择性的CX
4
与烯烃的加成反应,并通过实验和理论计算揭示了不对称催化中未曾研究过的反应机理。该反应原理也可能适用于其他对映选择性原子转移的自由基加成反应。该工作发表在
Angew. Chem. Int. Ed.
上,实验部分由德州大学西南医学中心Joseph Ready课题组的
陈波
(Bo Chen)博士完成,计算模拟部分由刘鹏课题组的博士研究生
方成
(Cheng Fang)完成。
该论文作者为:
Dr. Bo Chen, Cheng Fang, Prof. Peng Liu, Prof. Joseph M. Ready
原文(扫描或长按二维码,识别后直达原文页面):

Rhodium-Catalyzed Enantioselective Radical Addition of CX
4
Reagents to Olefins
Angew. Chem. Int. Ed.
,
2017
,
56
, 8780, DOI: 10.1002/anie.201704074
导师介绍
Joseph Ready
http://www.x-mol.com/university/faculty/3174
刘鹏
http://www.x-mol.com/university/faculty/1732


本文版权属于
X-MOL
(x-mol.com),未经许可谢绝转载!欢迎读者朋友们分享到朋友圈or微博!
长按
下图
识别图中二维码
,轻松关注我们!
点击“
阅读原文
”,查看
所有收录期刊






