
众所周知,PacBio测序技术是以其超长的测序读长而惊艳于世,无论是基因组的组装还是转录异构体的发现,片段读长越长对后续的分析越有利,好比我们玩拼图游戏一样,同一幅画,拼图方块越大越少越好拼,反之亦然。如图一所示,有一半的reads读长是大于20Kb的,最长的reads甚至超过60Kb。借助于超长的序列读长,对于转录组的研究来说,所获得的数据无需组装,一条reads就可以轻松跨过全长的转录本,极大的简化了后续的分析流程。这在第一期中已经说明,这里便不再赘述。
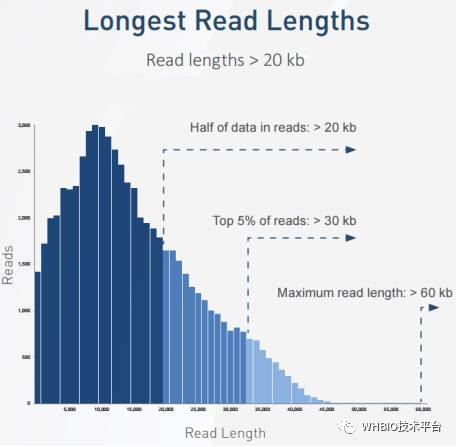
图一 PacBio RSII平台的reads长度分布图
为什么PacBio会有如此长的读长,而且Iso-Seq所测得的数据无需组装呢?除了Iso-Seq技术本身很牛之外,在文库的制备过程中,三代和二代也不尽相同。那接下来我们就好好捋一捋,弄个明白。图二显示了Iso-Seq和RNA-Seq的转录组建库流程差异,对于三代Iso-Seq测序而言,mRNA是无需片段化处理的,直接在逆转录酶的作用下合成第一链全长cDNA,接着通过聚合酶合成得到全长的双链cDNA文库,用于后续测序文库的构建,最后上机测序。由于PacBio的超长读长,最终所测的数据包含了整个全长cDNA的信息,减免了后续数据组装的流程,尤其减少了后期组装引入的偏差,数据的准确性更可靠。而反观RNA-Seq测序,原始mRNA首先需要经过片段化处理,然后在随机引物和逆转录酶的作用下合成得到片段化的单链cDNA,进而在聚合酶的作用下合成得到短的双链cDNA。由于二代测序读长本身较短,同时mRNA样本又经过了片段化处理,所以最后下机的数据是短而散的,后续需要先对数据进行组装才能用于其他分析。但是在组装的过程中,鉴于每种软件都有自己不同的算法,最后得到的结果也会有各种偏差。
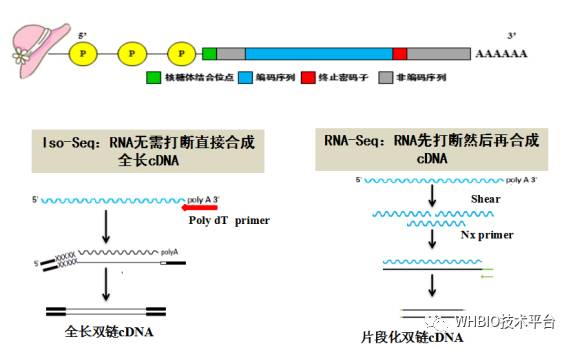
图二Iso-Seq全长转录组测序与RNA-Seq转录组测序的cDNA文库构建流程图
当然Iso-Seq除了建库时样本无需进行片段化处理以外,在逆转录时与RNA-Seq也有很大的差异,正是这个差异导致Iso-Seq测的是全长的cDNA。那全长cDNA又是怎么合成的呢?您请往下看。

图三 全长cDNA合成流程示意图
图三是用Clontech的SMART技术合成的全长cDNA,它的原理如下:以单链mRNA为模板,开始在polyA 尾部进行合成,当逆转录酶到达mRNA模板末端时,它的末端转移酶活性会自动在新合成的cDNA末端添加几个胞嘧啶(dC)。并作为合成寡核苷酸的结合位点与SMART II A Oligo末端的鸟嘌呤(dG)进行互补,然后反转录酶会自动转换模板继续延伸cDNA单链,直到SMART II A Oligo的末端,为第二链合成和后续PCR扩增产生通用的3'序列(如图黑色部分)。利用已知的引物序列可以方便地进行PCR,只有同时具备这两个结合位点的序列(如图黑色部分)才能够进一步合成得到双链cDNA。最后用其进行文库构建,上机测序,就得到一条一条全长的序列。
当然事事不会如此完美,对于5’端降解的mRNA,这款逆转录酶是识别不了的,只要他不脱离模板或者提前终止活性,他就会将其反转录并合成单链的cDNA。如果前期RNA的质量不佳,完整性不高,那纵使我们技艺再精,测序读长再长,最终得到的结果也会有很多非全长的转录本,毕竟巧妇难为无米之炊,上神最终也难逃应劫。
所以归根结底,测序的数据质量除了与实验设计、实验操作有关以外,最重要的还是样本的质量,这也是为什么PacBio测序一直对样本质量要求很高,因为我们要表里如一,这样才是最真实,也是最可靠的。

武汉生物技术研究院位于东湖国家自主创新示范区武汉光谷生物城内,成立于2009年8月29日,由湖北省委、省政府整合武汉大学、华中科技大学、华中农业大学、中科院武汉分院等高校、科研院所的优势资源组建而成的新型的科研/产业型研究院,其下设立的技术平台部已搭建以第三代测序为核心的基因测序分析,生物质谱检测及分析,生物核磁分析及筛选,细胞分选及显微成像,细胞/化合物高通量筛选,高性能运算与生物信息分析,生物发酵及制备,蛋白纯化及晶体解析共计八大设备平台,以及以基因组学、转录组学、表观组学、蛋白组学、代谢组学为支撑的综合性组学技术中心。近四年来,累计服务全国超过400个企事业单位、高校、科研院所,年均检测样本数超过15000个,服务客户超30000人次。承担国家十一五,十二五重大新药创制科技重大专项,国家病毒病防治重大专项等多项重大课题。










