本期介绍一个高通量测序质控的工具包:RSeQC包,它提供了一系列有用的小工具能够评估高通量测序尤其是RNA-seq数据.比如一些基本模块;
检查序列质量
,
核酸组分偏性
,
PCR偏性
,
GC含量偏性
,还有RNA-seq特异性模块:
评估测序饱和度
,
映射读数分布
,
覆盖均匀性
,
链特异性
,
转录水平RNA完整性
等。
安装:
RSeQC
是依赖于python的,直接使用
pip
进行安装:
pip install RSeQC
输入数据格式:
RSeQC接受4种文件格式:
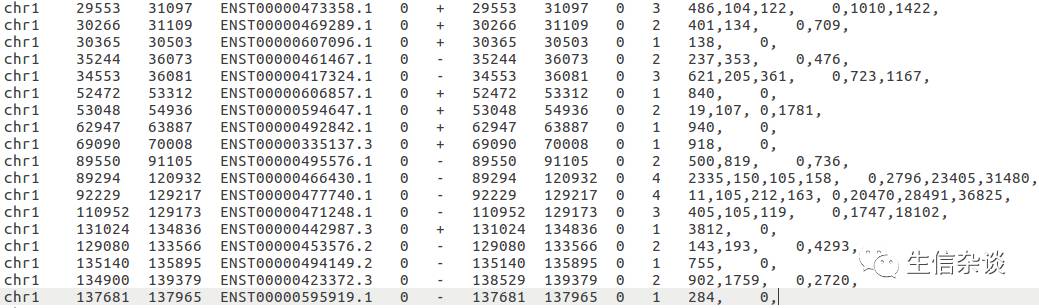
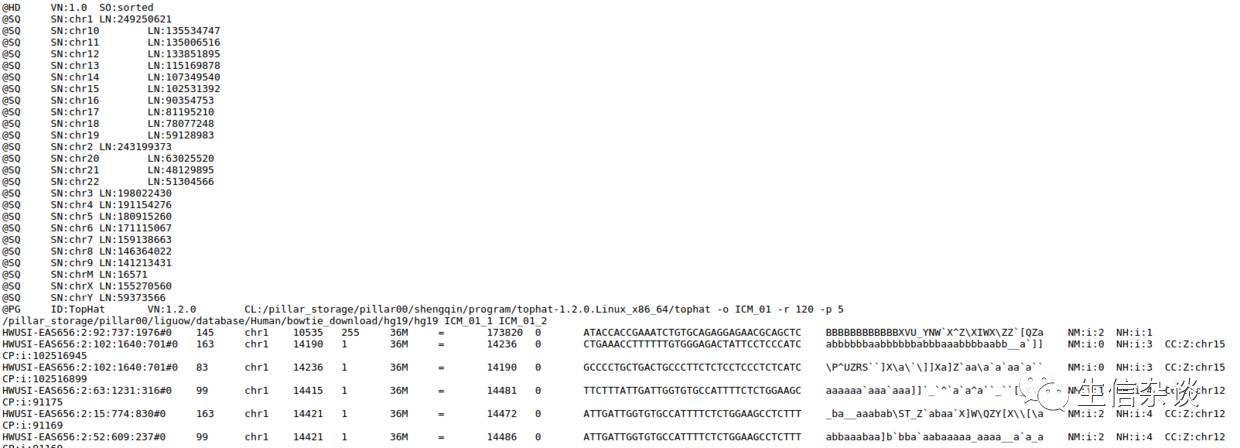
使用方法:
最新版本的
RSeQC(2.6.4)
包含以下一些模块,每个模块都可以单独调用进行分析:

我们一个一个来看:
bam2fq.py:
将
BAM
或
SAM
格式的文件转为
fastq
格式.(这个感觉一般用不到)
bam2wig.py:
将BAM文件转为
wig
/
bigwig
格式的文件.(这个在作图尤其是信号图的时候很有用.)如果需要转为
bigwig
格式,则需要UCSC的
wigToBigWig
工具,下载地址:
http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/wigToBigWig
bam_stat.py:
对比对结果文件
BAM
或
SAM
文件进行统计.其实
samtools
里也有类似工具.结果如下所示:
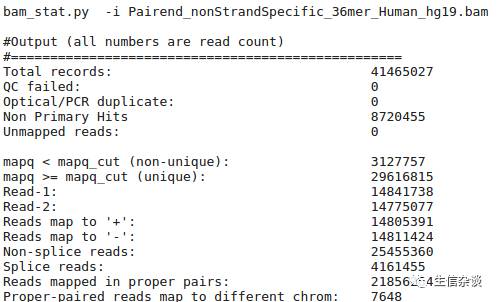
统计结果包括:
总比对记录
,
PCR重复数
,
Non Primary Hits
表示多匹配位点.
不匹配的reads数
,
比对到+链的reads
,
比对到-链的reads
,
有剪切位点的reads
等.
clipping_profile.py:
这个模块用于评估
RNA-seq
的
BAM
或
SAM
文件中的
有切除核苷酸的reads
情况.
clipped reads
有两种,一种是
soft-clipped
,即
reads
5’或3’不能比对到参考基因组;另一种是
hard-clipped
,即
reads
5’或3’不能比对到参考基因组并且被剪切.
这个模块会生成
.r
格式的作图脚本以及
.pdf
格式的报告文件以及
.xls
的数据文件.
例如双端测序
clipping profile
图如下:
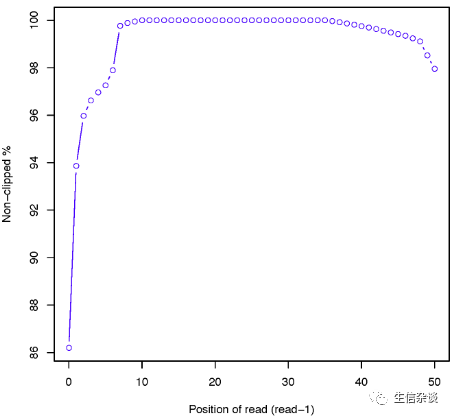
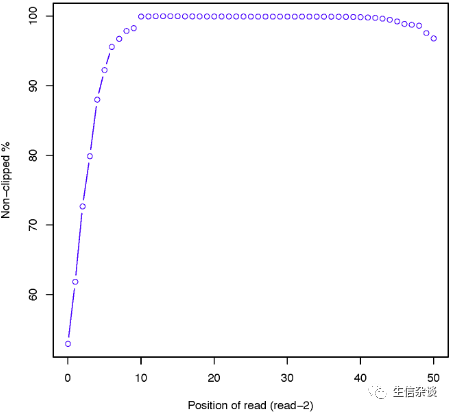
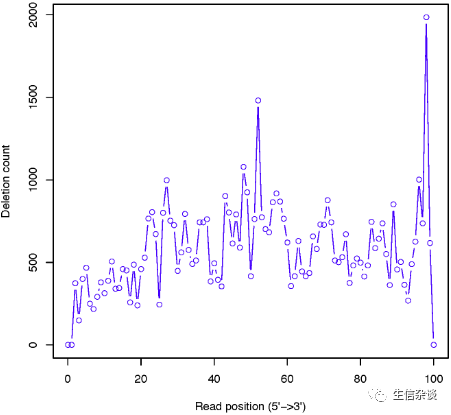
deletion_profile.py:
也就是
reads deletion
位点的分布.
divide_bam.py:
随机分割
BAM
文件(m个比对结果)为n个文件,每个文件包含
m/n
个比对结果.
FPKM_count.py:
根据
read count
和
gene注释文件
(bed12格式)计算每个基因的
FPM
(fragment per million)或者
FPKM
(fragment per million mapped reads per kilobase exon).
结果类似下面的:
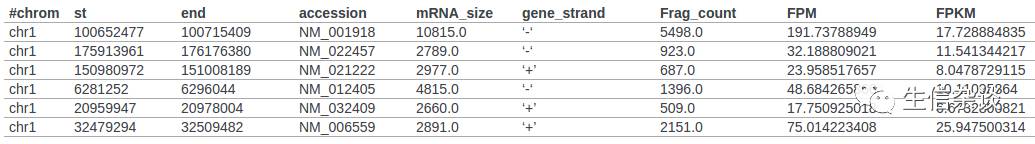
geneBody_coverage.py:
计算RNA-seq
reads
在基因上的覆盖度.
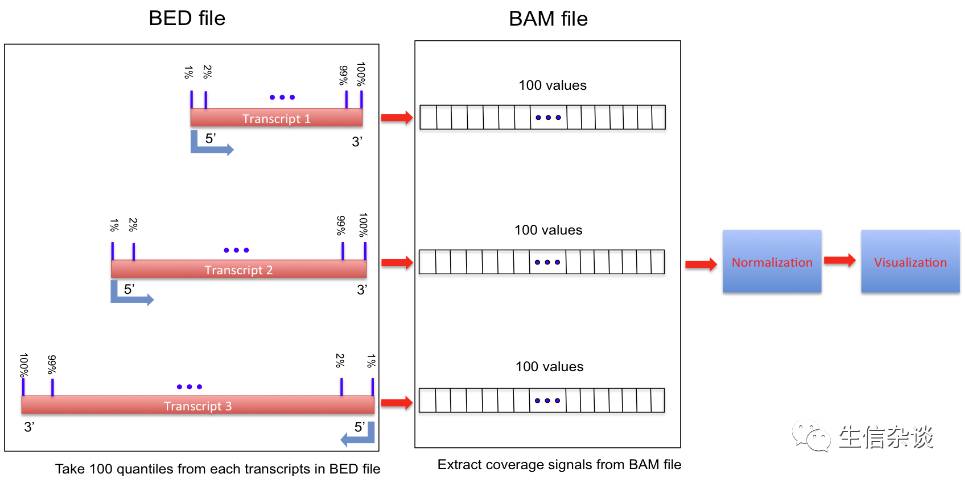
结果如下:
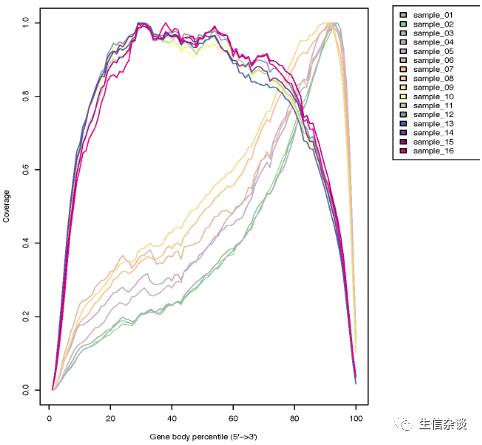
如果是大于3个的样本,则还会生成热图:
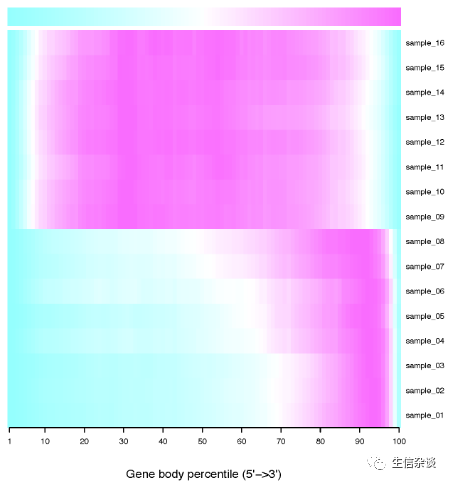
geneBody_coverage2.py:
功能和上面的
geneBody_coverage.py
一样,但输入的是
bigwig
格式文件
infer_experiment.py:
-
这个模块用来”猜”RNA-seq的相关配置信息,针对
链特异性测序
,通过
reads的链型
与
转录本的链型
来评估
reads
是哪一条链的.
-
reads的链型
是通过比对结果得到的,
转录本的链型
是铜鼓注释文件得到的.
-
对于
非链特异性测序
,
reads的链型
与
转录本的链型
是没有关系的.
-
对于
链特异性测序
,
reads的链型
与
转录本的链型
是有很大关系的.通过下面的3个例子说明.
-
在测序前你并不需要知道RNA-seq是不是链特异性的,就当他们是非链特异性的,这个模块可以”猜”到”reads”是哪条链的.
对于双端
RNA-seq
,有两种方法来确定reads在哪条链(如illumina ScriptSeq protocol):
(1).
1++,1–,2+-,2-+ :
说明:
1
和
2
表示
read1
和
read2
,第一个
+/-
表示read map 到哪条链,第二个
+/-
表示这个read 所match的基因在哪条链.
那这个
1++,1–,2+-,2-+
就表示
reads
所match的链和其所在gene的”+/-“是一样的,也就是reads的链型与其基因的链型一样,是不独立的.
(2).
1+-,1-+,2++,2– :
这个就表示
reads
所match的链和其所在gene的”+/-“是不一样的,也就是reads的链型与其基因的链型是不独立的.
对于单端测序:
(1).
++,– : 表示
read链型
与其所match的
gene链型
一致.
(2).
+-,-+ : 表示
read链型
与其所match的
gene链型
不一致.
举三个例子说明:
例一:

解释:
总
reads
数的
1.72%
被映射到基因组区域,我们无法确定这样的
gene的链型
(比如这个区域两条链都转录)。 对于剩余的
98.28%
(1 - 0.0172 = 0.9828)的reads,一半是“1 ++,1-,2 + - ,2- +”
reads
与
gene
链型一致的,而另一半是“1 +1 - +2++,2-”不一致的。 我们得出结论,
这不是一个链特异性的测序
,因为
reads链型
不依赖于
gene链型
.
例二:
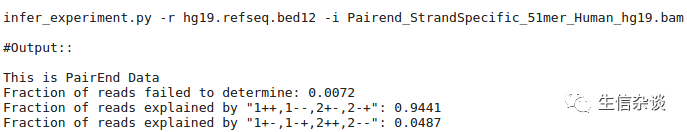
解释:
0.72%
是不能确定链型的.
94.41%
的
reads
与
gene
链型一致的.仅有
4.87%
是不一致的.我们得出结论,
这是一个链特异性的测序
,因为
reads链型
依赖于
gene链型
.
例三,这是个单端测序的例子:
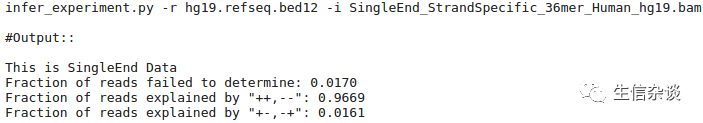
解释:
reads链型
依赖于
gene链型
,这个是链特异性测序.
inner_distance.py:
针对双端测序,计算
read pairs
的
内部距离
或者
插入距离
,关于
插入距离
是什么,看下图:
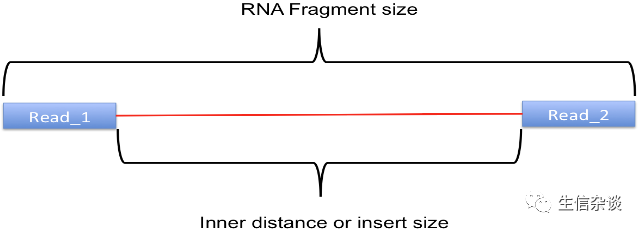
插入距离
D_size
计算公式:
D_size = read2_start - read1_end
但是不同条件下计算方法是不一样的:
-
paried reads
比对到同一个外显子:
插入距离
= D_size
-
paried reads
比对到不同外显子:
插入距离
= D_size - intron_size
-
paried reads
比对到非外显子区域(如内含子或者基因外区域):
插入距离
= D_size
-
如果两个
fragments
重叠则
插入距离
可能为负.
结果举例:
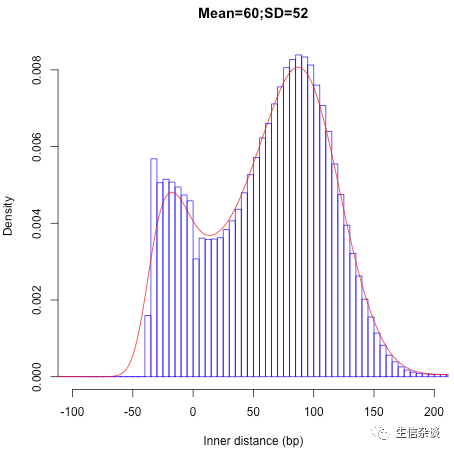
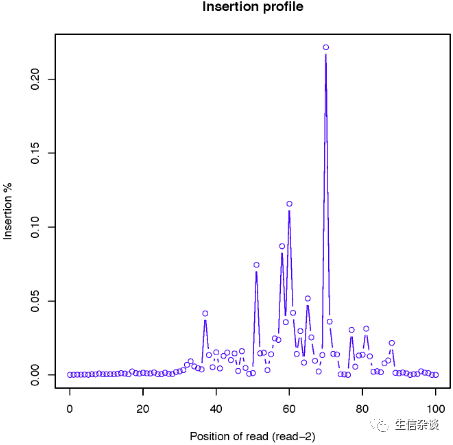
insertion_profile.py:
计算reads上被插入核苷酸的分布.
结果举例:
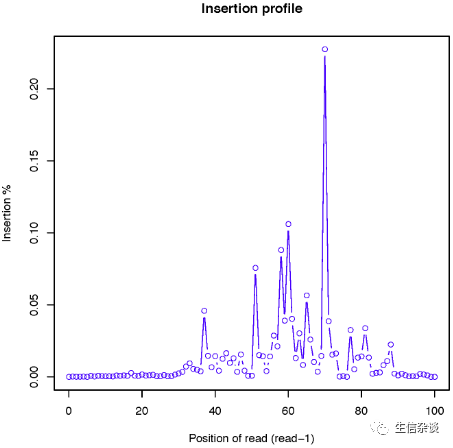
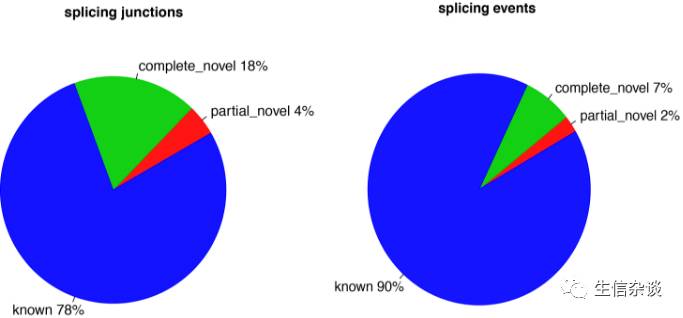
junction_annotation.py:
输入一个
BAM
或
SAM
文件和一个
bed12
格式的参考基因文件,这个模块将根据参考基因模型计算剪切融合(splice junctions)事件.
-
splice read
: 一个RNA read,能够被剪切一次或多次,所以100个
spliced reads
能够产生>=100个剪切事件.
-
splice junction:
多个跨越同一个内含子的剪切事件能够合并为一个
splicing junction
.
junction 有三种:
-
已经被注释的.
5'剪切位点
和
3'剪切位点
已经被注释.
-
全新的.
-
部分是新的.
结果示例:
junction_saturation.py:
在剪切位点分析时首先检查当前的
测序深度
是否是足够深的.对于一个已经注释的物种,在某个组织中基因的数量是一定的,所以剪切位点数量也是一定的.可以从参考基因模型(
bed12
格式的文件)可以提前看出
splice junctions
的数量. 一个饱和的RNA-seq能够发现所有已经被注释的
splice junctioons
,否则下游剪切位点分析将会出现问题因为将有较少的
splice juncitons
将发现不了.这个模块从这个测序结果(
SAM
或
BAM
文件)中进行重抽样,从5%,10%,15%,…,到95%的饱和度来检查每个阶段的
splice junctions
并与参考基因组进行比较.
结果示例:
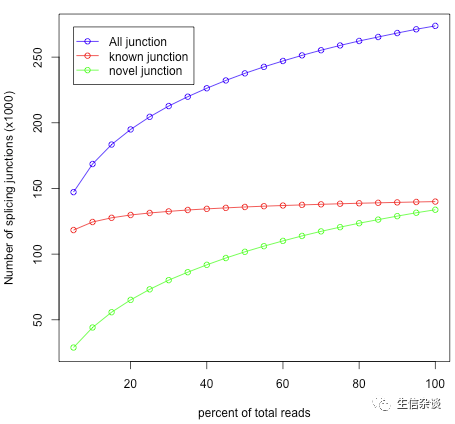
解释:
在这个例子中,每个饱和度下的测序深度的
known junctions
基本都是一致的(
红线
).因为大部分的剪切位点我们基本都发现了.即便加深测序深度也不会发现跟多的
known junctions
,仅会加深junciton的覆盖度(如junction被更多的reads覆盖.).然而当前测序深度(100%的reads)对于发现新的juncitons是不够的(
绿线
)
mismatch_profile.py:
计算reads的不匹配位点的分布.
结果示例:
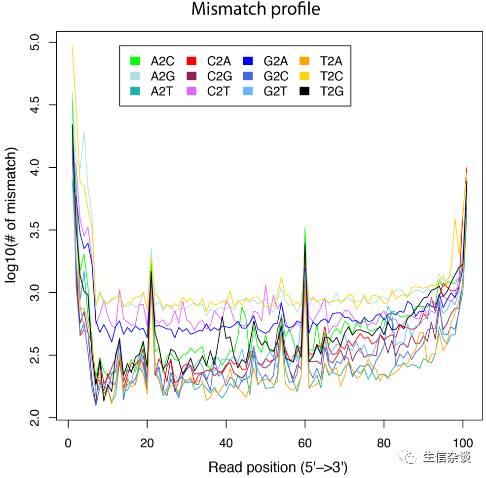
上图可以看出5’和3’的不匹配位点最多,这是由于测序本身所决定的.
normalize_bigwig.py:
可视化
RNA-seq数据结果是最直接并且高效的质控方式.但在可视化之前我们要保证所有样本的数据是可比较的,这就需要进行
归一化
.信号值文件’wig’或
bigwig
文件主要由两个因素决定:
(1)
总reads数,
(2)
read长度.因此,如果两个样本的read长度不一样但仅对”总reads数”归一化是有问题的.这里我们将每个
bigwig
文件归一化到相同的
wigsum
值.
wigsum
是对基因组信号值的汇总,例如:
wigsum
=100,000,000等价于
1百万个100nt的reads或2百万个50nt的reads的覆盖度
. 结果生成
wig
格式的文件.
overlay_bigwig.py
这个模块让我们操作两个
bigwig
文件.可以采取的操作有:
信号值相加
,
取均值
,
相除
,
每个信号值+1
,
求最大值
,
求最小值
,
相乘
,
相减
,
求几何平均数
.
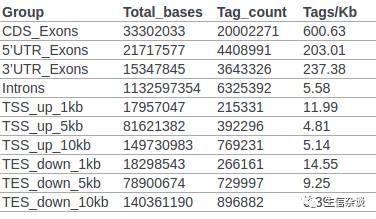
read_distribution.py:
这个模块根据提供的
BAM/SAM
文件和
bed12格式的
gene模型文件就按比对上去的
reads
在基因组上的分布情况,比如在
CDS exon
,
5'UTR exon
,
intro
,
基因间区域
的
reads
分布.
结果示例:
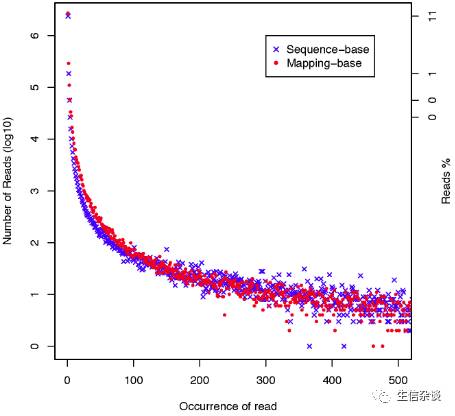
read_duplication.py:
两种用于计算重复率的策略:
(1)
基于序列的,完全相同序列的reads被视为重复的reads.
(2)
正好map到同一个基因组位置的reads被视为重复reads. 对于
splice reads
,map到同一位置并且以相同方式剪切的视为是重复reads.
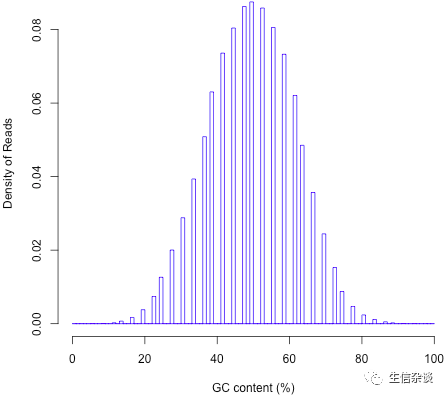
read_GC.py:
计算reads的GC含量分布.
read_hexamer.py:
计算6mer的频率.
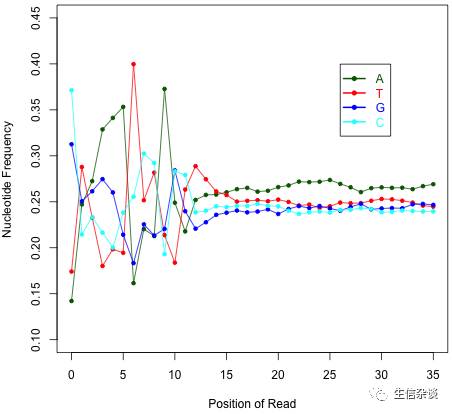
read_NVC.py:
这个模块有用来检查核苷酸的碱基组成偏好性.由于随机引物的影响,
reads 5'端
开始会有某些模式过表达.这种偏好性能够被
NVC
(Nucleotide versus cycle)画出.
理想状态下, A%=C%=G%=T%=25%
.
结果示例:
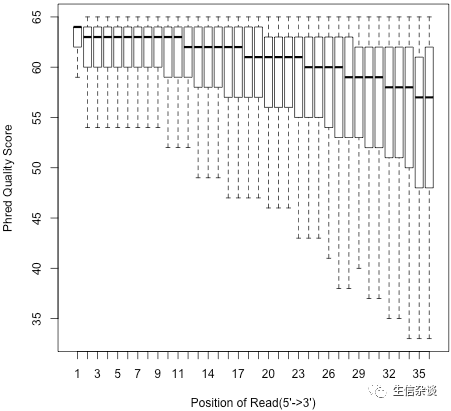
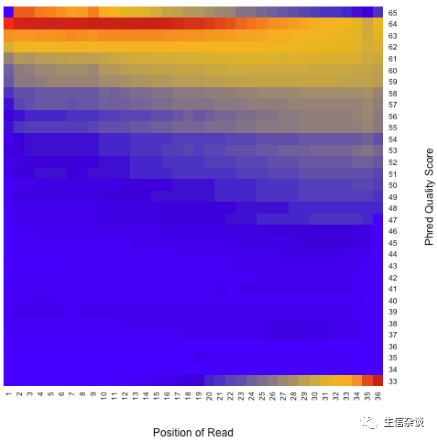
read_quality.py:
可视化
reads
每个位置的测序质量.
结果示例:
RNA_fragment_size.py:
在map后计算每个gene上的
fragment
的大小,包括:每个gene上所有的
fragment
的均值,中位数,方差.
结果示例:

RPKM_count.py:
这个模块在最新版里已经被废弃,如果想使用可以翻看以前的版本.
RPKM_saturation.py
任何样本统计(
RPKM
)的精度受样本大小(
测序深度
)的影响;
重抽样
或
切片
是使用部分数据来评估样本统计量的精度的方法. 这个模块从总的
RNA reads
中重抽样并计算每次的
RPKM
值.
通过这样我们就能检测当前测序深度是不是够的(如果测序深度不够RPKM的值将不稳定,如果测序深度足够则RPKM值将稳定).
默认情况下,这个模块将计算20个
RPKM
值(分别是对个转录本使用5%,10%,…,95%的总
reads
).
在结果图中,Y轴表示
“Percent Relative Error”
或
“Percent Error”
.用来表示当前样本量下的
RPKM
与实际表达量的偏差.计算公式如下:
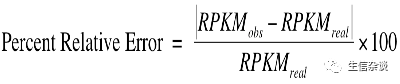
结果示例:
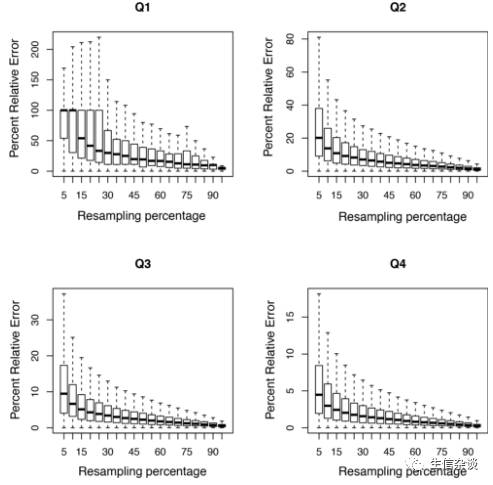
说明:Q1,Q2,Q3,Q4是按照转录本表达量4分位分开的.Q1表示的是表达量低于25%的转录本,以此类推.
可以看出:
随着样本量升高,
RPKM
与实际值的偏差也在降低.而且转录本表达量越高这种趋势越明显(Q4最明显).
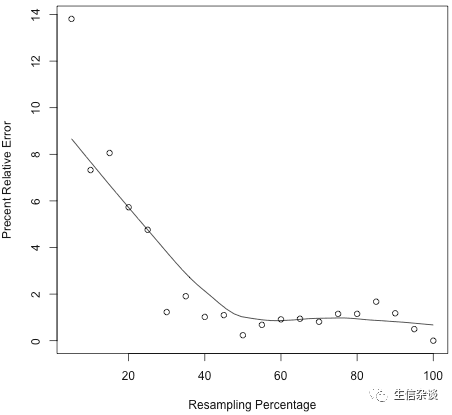
可以看出,样本量50%之后线条已经趋于平缓,也就是说对于转录本定量来说,当前测序深度是足够的.
spilt_bam.py:
根据提供的
bed12
注释文件和
BAM
文件拆分为以下三个文件:
-
XXX.in.bam:
包含map到外显子趋于的reads.
-
XXX.ex.bam:
包含map不到外显子趋于的reads.
-
XXX.junk.bam:
质控失败或者没有map上去的reads.
split_paired_bam.py:
将一个
双端测序
的
BAM
文件拆分为两个
单端测序
的
BAM
文件.
tin.py:
这个模块用来在转录本级别计算RNA完整性TIN (transcript integrity number)值.
结果示例:
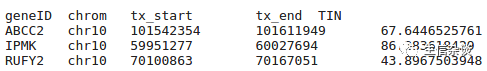
每个模块的详细参数请点击左下角"阅读原文"查看官方文档.
更多原创精彩视频敬请关注
生信杂谈:






