透皮贴剂是一种运用经皮给药系统(Transdermal Delivery System,TDS)或经皮治疗系统(Transdermal Therapeutic System,TTS)释放药物,从而产生治疗作用的制剂。我国国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)于2020年12月23日颁布的《化学仿制药透皮贴剂药学研究技术指导原则(试行)》[1],以及《中华人民共和国药典》(以下简称《中国药典》)2020年版四部贴剂通则[2]将透皮贴剂定义为用于完整皮肤表面并且能将药物输送透过皮肤进入血液循环系统起全身作用的贴剂。
透皮贴剂使药物以恒定或接近恒定速度通过皮肤(表皮、真皮和皮下组织)进入人体血液循环,可避免肝脏首过效应及在胃肠道的降解。透皮贴剂的特点:可保持血药浓度稳定在有效的治疗浓度内,避免口服等给药引起血药浓度的峰谷现象,降低了毒副作用;使用方便,能够克服口服给药时儿童不愿吃药、老人吞咽能力差的弊端;在用药过程中出现问题可随时停药,使用灵活;尽管有如此多的优点,但是由于皮肤是限制体外物质吸收进入体内的生理屏障,且大多数药物透过该屏障的速度都很慢,所以,透皮贴剂通常需要给药几小时才能起效,还有一些药物不能达到有效治疗浓度,且部分对皮肤有刺激性和致敏性的药物不宜被设计成透皮贴剂。
在新药开发的过程中,临床试验耗资和耗时基本均占整个周期的60%~80%。目前,我国透皮贴剂的发展还不成熟,其临床研究相关的具体技术要求尚不完善,仅有2022年1月7日CDE发布的《改良型新药调释制剂临床药代动力学研究技术指导原则》。因此,节约研发成本、加快我国透皮贴剂的上市进程,科学、合理地设计透皮贴剂的临床研究将是趋势。本文就欧美关于透皮贴剂的临床研究的要求及实践进行综述,并结合案例进行讨论和分析。
对于新分子实体来说,美国505(b)(1)和欧盟Full Application申报路径往往要求申请人提交完整的临床研究数据[3-4]。通常,申请人需在少量的健康志愿者身上开展Ⅰ期研究以获得药物的基本安全性数据,随后在小部分受试者身上开展Ⅱ期研究以进一步获得安全性、对目标适应症的有效性数据,而Ⅲ期研究则重点考察药物的安全性和有效性。欧盟《调释制剂的药代动力学和临床研究评价》(Guideline on Pharmaco Kinetic and Clinical Evaluation of Modified Release Dosage Forms)要求当一种新分子实体被开发成透皮贴剂时应开展体内和体外研究,药代动力学应包括单剂量和多剂量,同时考察剂量比例。此外,还应考察贴片的皮肤刺激性、致敏性、光毒性及黏附性。评估黏附性时还应考虑外部因素的影响(如外热或防晒霜),若产品的预期患者中存在老年人,应在皮肤状况与预期患者相似的受试者中进行以上皮肤测试[5]。
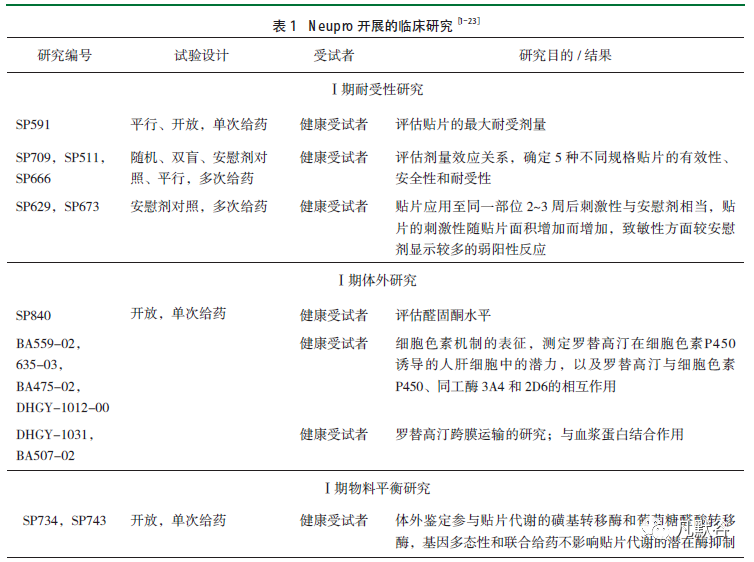
以欧美批准的罗替高汀贴片为例(商品名:Neupro),其临床研究主要包括了健康受试者和患者的药物代谢动力学(Pharmacokinetic, PK)和药效学(Pharmacodynamic, PD)研究,例如代谢研究、物料平衡、内在因素、外在因素、耐受性、生物等效性研究等,并考察了长期安全性。在有效性和安全性方面,关键的Ⅲ期研究SP512、SP513及SP650均与SP506显示出了相似的疗效结果,且贴敷部位对临床疗效没有影响。但整个临床研究期间没有考察光毒性和外热的影响。
透皮贴剂创新药临床试验的关键在于证明药物的安全性和有效性,临床药理学研究阶段应开展充分的单次和多次给药的PD、PK和药物相互作用研究。PK方面可考察产品的PK特性、吸收、代谢与排泄、年龄、性别、人种及特殊人群的PK差异、剂量比例以及不同贴敷部位的生物利用度,以及相关的体外研究,例如酶和受体的作用。PD方面可考察人体对新药的耐受剂量、不良反应、剂量-效应关系以及贴片的黏附性、刺激性和致敏性,Ⅱ期研究应考察剂量范围、为Ⅲ期临床提供合适的给药方案并评估患者短期内的不良反应,初步考察有效性和安全性。Ⅲ期研究可开展安慰剂或活性对照试验确证产品的安全性及有效性,并评估长期安全性,见表1。