生信草堂
将会与更多的优秀微信公众号合作,把最优秀的微信推文呈现给大家,希望可以帮助读者更多的了解生信技术,培养和提高读者的生信分析能力!
号外,号外,号外
你想和生信分析大神做好朋友么?
你想认识更多爱好生信分析的小伙伴么?
你想让自己的生信分析走上快车道么?
那就赶快加入我们的生信交流微信群吧!
正确加入我们的模式是:添加我们的微信
Edison686868
或者
mly-1800
为好友,标注“
加入生信草堂交流群
”
生信大讲堂
微信公众平台,作为浙江大学农业与生物技术学院作物所研究生第七党支部“生信大讲堂”生物信息系列讲座的重要线上平台,结合学科特色,坚持以“四讲四有”中“讲奉献有作为”作为活动核心价值观,为广大科研工作者提供了学习生物信息学相关学科知识的资源及平台。
~戳
这里
生信大讲堂
公众号原文,请多关注哦~
LD decay在群体遗传研究中是最重要和常见的分析内容之一,是衡量LD水平的重要指标。特别在自花授粉的作物中,LD衰减不仅能够反映作物驯化和育种历史,还能显示基因流现象、选择区域等。
传统LD处理软件haploView可以用来计算LD、单倍型等,但上百万snp标记的计算会消耗大量资源,本地运行往往会由于运行内存不足而崩溃。PopLDdecay是一种简单有效的LD衰减分析软件,可计算和绘图,以压缩包形式输入输出内容,相比其他软件,更适合大数据量的计算和分析。

https://github.com/BGI-shenzhen/PopLDdecay
$tar -zxvf PopLDdecay3.29.tar.gz #解压
$cd PopLDdecay3.29
$cd src
option1:
$make #安装
$make clean
option2:
$sh make.sh #安装
a.SNP数据(VCF文件)
用法:
PopLDdecay [options] -InVCF *.vcf.gz -OutStat *
Options & Parameters:
-InVCF
: 输入gzip压缩过的vcf格式的snp数据
-InGenotype
:输入基因型格式snp数据
-OutStat
:标准输出压缩包的前缀
-SubPop
:亚群体名称列表(与snp文件个体名称对应,单行制表符分隔)
-MaxDist
:两个snp之间的最大计算距离(kb),默认300kb
-MAF
:[过滤snp]最小等位基因频率,默认0.5%
-Het
:[过滤snp]Max ratio of het allele [0.88]
-Miss
:[过滤snp]最大snp缺失率,默认0.25
-OutFilterSNP: 输出过滤后的snp数据
-OutPairLD
:输出成对snp计算得到的LD信息,0/2不输出,1/3/4输出简要信息,5输出全部信息,默认0,即不输出
-Methold
:选择计算的算法,1是Low MEM,2是May Big MEM,默认是1(此处有bug,软件参数中单词拼错了多一个l)
-help:打印帮助
b.Plink文件(.ped/.map)
第一步:将plink文件转换成基因型格式文件(Genotype)
perl plink2genotype.pl -inPED in.ped -inMAP in.map -outGenotype out.genotype
第二步:执行PopLDdecay
PopLDdecay -InGenotype out.genotype -OutStat LDdecay
第一种情况,只是为了单纯的看群体的LD情况,不考虑群体内部。
PopLDdecay [options] -InVCF *.vcf.gz -OutStat *
第二种情况,群体内分成若干个亚群体,比较亚群体之间的LD差别
PopLDdecay -InVCF
-OutStat
-SubPop A.list
在第一种情况用法之上,添加-SubPop参数,将同一亚群体的sample名称放在一个list中(与snp文件个体名称对应,单行制表符分隔),计算n个亚群体的LD则需要有n个list,执行上述命令n次。
单个群体:
perl Plot_OnePop.pl -inFile LDeecay.stat.gz -output Fig
options¶meters:
-inFile
: 计算的结果文件(.stat.gz)
-output
: 输出文件前缀。
多个群体:
perl Plot_MutiPop.pl -inList Pop.ReslutPath.list -output Fig
option & parameters:
-inList Pop.ResultPath.list: 亚群体LD计算结果路径及亚群体名称(一个群体一行,路径与名称之间制表符分隔)
染色体会有很多条,最后想得到全基因组范围内的LD衰减情况,因此需要将所有的染色体信息合并到一起:
方法1:
每条染色体得到LD计算结果,将每条染色体结果文件路径写在一个list中。然后执行命令:
perl Plot_OnePop.pl -inList Chr.ReslutPath.List -output Fig
方法2:
计算LD之前,将所有的snp文件合并在一起:
sed -i ‘/^#/d’ChrX.vcf
##除第一条染色体保留头文件外,其余染色体去除头文件信息
cat *.vcf > merged.vcf
##合并所有的vcf文件,然后再计算LD和绘图。
最后的结果文件是LD衰减图,pdf和png两个格式。如果对绘图使用的颜色等不满意或色彩差异不明显,可以修改绘图脚本(Plot_MutiPop.pl、Plot_OnePop.pl)中的颜色设置等。
查看具体的r2情况,可以将LD计算结果文件(.stat.gz)解压后查看,内容主要是不同距离对应的r2平均值等信息。可以在结果文件中找到r2值衰减到最大值一般时对应的距离,即为衡量LD水平的指标。
软件的不足之处很明显,除了平均后的r2和距离,你可能看不到其他信息,比如具体两个座位之间的r2和距离。因此软件只适用于全基因组或染色体范围内获得整体LD信息。
所以,PopLDdecay是一个只做全基因组或染色体整体LD衰退情况的软件,想要获取具体LD信息,出门左转见Haploview、TASSEL等软件。
Haploview是一个做单倍型、LD分析的软件。
〇
下载地址:
https://www.broadinstitute.org/haploview/downloads
〇
要求:
JRE(Java运行环境)5.0 Update 22以上
Windows版本可以下载安装器HapInstall.exe;
Mac/Unix/Linux版本直接下载JAR,然后再命令行直接运行:
java -jar Haploview.jar [options]
Windows推荐直接下载JAR文件,直接双击运行或命令行调用均可以。

HaploView功能:
1.连锁不平衡LD与单倍型分析
2.单倍型人群频率估算
3.SNP与单倍型关联检验
4.关联显著性排列检验
5.从HapMap上下载基因型信息
6.PLINK全基因组关联结果可视化
支持的输入文件格式有Linkage Format、Haps Format、HapMap Format、HapMap PHASE、HapMap download和PLINK Format。
Linkage Format格式文件(.ped)无头文件,列依次是Pedigree Name、Individual ID、Father ID、Mother ID、Sex、Affection Status、Marker Genotypes。
Marker位置信息文件(.info)分成2列,分别是Marker ID和位置。Hapmap Format文件可以在Hapmap网站上下载数据。
今天我们将hapmap文件转换成Linkage Format文件后再进行LD分析(其实可以直接导入hapmap文件,不过小编就是想用用刚学的python做一些简单的文本处理,哈哈哈哈)。
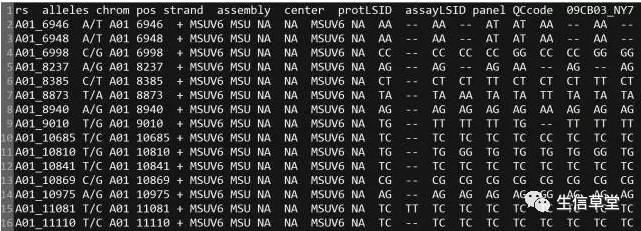
Hapmap文件格式如上,首先按照Linkage Format的要求,提取出Marker的位置信息(.info)。
###刚学python,别笑
###Python script
###hapmap_2_linkage_format_marker_info
input_file=input("INPUT FILE: ")
a=open(input_file,'r').readlines()
b=a[1:]
output=open('test.info','w')
for c in b:
d=c.split('\t')
e=[]
e.append(d[0]+'\t'+d[3]+'\n')
e=e[0]
output.write(e)
output.close()
处理hapmap文件,单独提取一个individual作为例子,保存到一个文本中,内容只有一列,但是有很多行(与.Info文件行数相同)。然后简单脚本处理,按照ped文件格式要求输出到新文件(.ped),比如将AGCT换成对应数字1234等,最后ped文件只有一行多列。

###刚学python,别笑
###Python script
input_file=input('INPUT FILE: ')
open_file=open(input_file,'r')
read_file=open_file.readlines()
ID=(read_file[0]).rstrip('\n')+'\t'
file_name=(read_file[0]).rstrip('\n')+'.ped'
out_file=open(file_name,'w')
snp=read_file[1:]
g=[]
for i in snp:
i=i.rstrip('\n')
g.append(i)
h=''.join(g)
m=''
for j in h:
if j=='A':
j='1'
elif j=='C':
j='2'
elif j=='G':
j='3'
elif j=='T':
j='4'
else:
j='0'
m=m+j+'\t'
ID='Test'+'\t'+ID+'0\t0\t0\t2\t'+m.rstrip('\t')+'\n'
out_file.write(ID)
out_file.close()
选择需要的群体将ped文件合在一起,与info文件命名相同后,导入。选定ped文件后,info会自动选择。默认距离超过500kb的marker之间不做分析,缺失率超过50%的SNP被去除。Loading的过程中其实在进行计算。
计算结果有4栏:LD Plot、Haplotypes、Check Marker、Tagger。
LD:
颜色由浅至深(白——红),表示连锁程度由低到高,深红色表示完全连锁,即r2=1。在方块上点击右键,可以看到连锁具体信息,包括R2、D’、LOD、基因型频率等。具体的r2计算值可以file中导出。
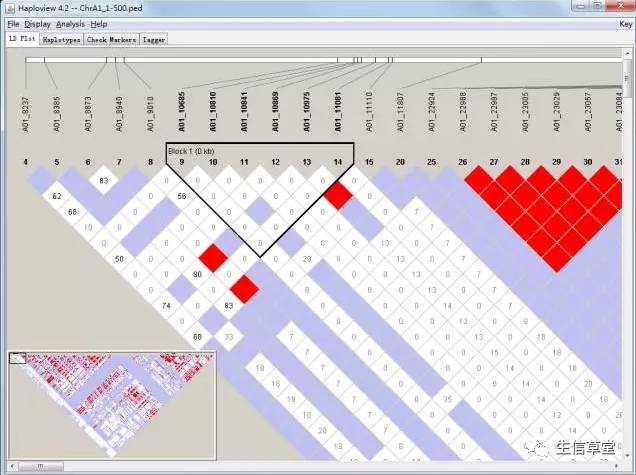
Haplotypes
:单倍型
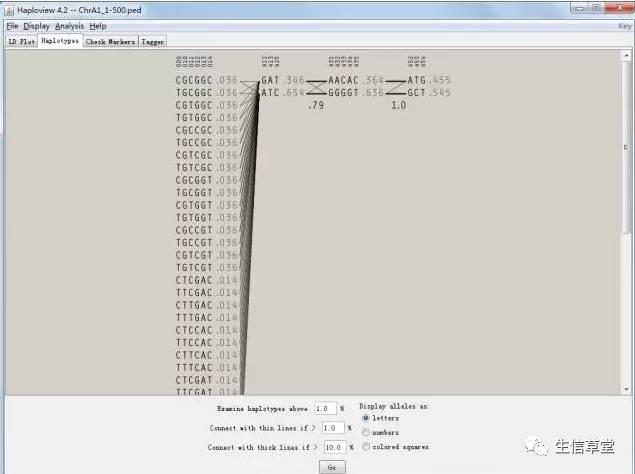
Check Marker:
对marker质量进行检查,比如缺失率,MAF等,可以自己设置筛选阈值。

Tagger
:可以进一步选择标签SNP
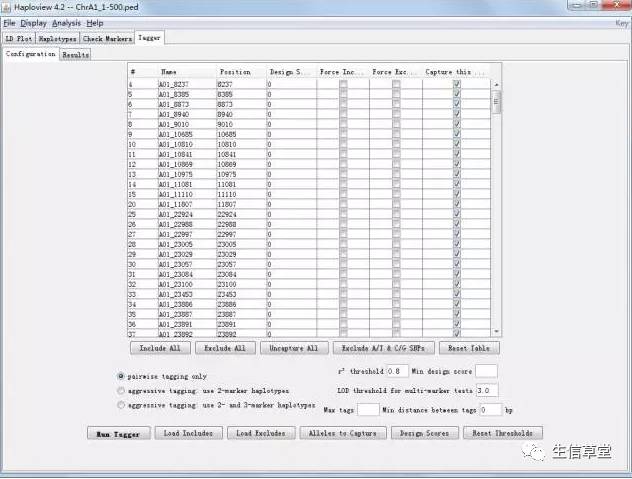
如果SNP数量有几十万或更多,直接默认运行程序很可能会java报错超过内存限制(java.lang.OutOfMemoryError或者GC overhead limit exceeded),需要在命令行添加参数运行,比如java -Xmx4096m _Xms2048m -jar Haploview.jar。
对于万级以下个marker,默认运行内存足够。使用Haploview做全基因组SNP的LD分析体验效果很差,并不推荐,但对于一般遗传标记(AFLP、SSR、RAPD等)还是值得一试的。
 长按关注生信草堂
长按关注生信草堂







