接下来我们对m
6
A抑制
notch1a
表达的分子机制进行了探索。经过m
6
A甲基化修饰的mRNA可以被不同的“读码器”识别并走向不同的命运,其中YTHDF2能够介导m
6
A甲基化mRNA的降解,因此也暗示着其可能在m
6
A对
notch1a
的抑制过程中发挥作用。为证明这一猜测,我们首先确认了ythdf2缺失能够导致造血干细胞产生缺陷(图4b和4c)。随后,我们将已报道的YTHDF2对m
6
A的识别位点进行突变,通过在细胞内的表达最终得到斑马鱼的YTHDF2-MU突变蛋白(含W445A、W499A和W504A三个点突变)。EMSA实验结果表明,相比于野生型YTHDF2蛋白,突变YTHDF2丧失了对m
6
A的识别及结合能力(图4a)。同时,在
ythdf2
缺失胚胎中的造血干细胞缺陷表型可以被
ythdf2
mRNA过表达回救,但是不能被
ythdf2-mu
mRNA过表达回救(图4b和4c)。以上结果说明YTHDF2对造血干细胞的调控作用依赖于其对m
6
A的识别。
为进一步证明YTHDF2对m
6
A所修饰目标基因的调控作用,我们进行了YTHDF2-RIP-Seq。结果发现在YTHDF2结合的RNA中有72.3%的基因同样能够被m
6
A修饰,进一步说明YTHDF2对m
6
A修饰RNA的特异性结合。随后我们从对照组和
ythdf2
缺失组Tg(
kdrl
:mCherry;
runx1-en-
GFP)胚胎中分选出内皮细胞并对其进行深度转录组测序。相关性分析发现,在m
6
A和YTHDF2共同的靶基因中,有包括
notch1a
在内的472个基因在
mettl3
和
ythdf2
缺失的胚胎中表达都有上调,说明它们是m
6
A和YTHDF2的共同目标基因。GO分析发现这些表达上调的基因主要富集在信号通路和血管发育中(图4d),这与我们之前对m
6
A靶基因的分析结果一致。接下来我们分析了YTHDF2对
notch1a
mRNA的结合。结果发现在m
6
A结合的位置也有YTHDF2的结合,而且RNA-Seq也发现
notch1a
在
ythdf2
缺失的胚胎中表达明显上调(图4e)。
为进一步证明YTHDF2对
notch1a
的调控作用,我们在分选的内皮细胞中通过qPCR检测
notch1a
基因的表达。同RNA-Seq结果相同,
notch1a
基因的表达在
ythdf2
缺失胚胎中显著升高(图4f)。同时,Western blot检测结果显示Notch1蛋白水平的表达在
ythdf2
缺失胚胎中有明显上调(图4g)。随后,我们还利用Notch信号的转基因鱼报告系Tg(
fli1a
:EGFP;
tp1-
dsRed)来检测Notch信号的活性。共聚焦荧光显微镜观察结果显示,Notch信号激活的内皮细胞数目在
ythdf2
缺失胚胎中有显著上调(图4h和4i)。为阐明YTHDF2调控
notch1a
表达是否是通过其介导的mRNA降解途径,我们利用转录抑制剂actinomycin D从24 hpf开始处理对照组和
ythdf2
缺失组胚胎,并分别在处理后4小时、8小时提取RNA进行qPCR检测。结果显示,相比于对照组中actinomycin D处理后
notch1
mRNA的快速降解,我们发现在
mettl3
或
ythdf2
缺失胚胎中,
notch1a
mRNA在actinomycin D处理后能够维持较高水平(图4j)。为进一步阐明YTHDF2对
notch1
mRNA稳定性的调控依赖于其特定位点的m
6
A甲基化修饰,我们进行了单碱基分辨率的m
6
A-miCLIP-Seq,并发现
notch1a
mRNA上存在功能性的m
6
A修饰位点,并可以介导造血干细胞的表型。由此可知,YTHDF2能够通过促进mRNA降解来抑制
notch1a
的表达,进而调控造血干细胞的产生。
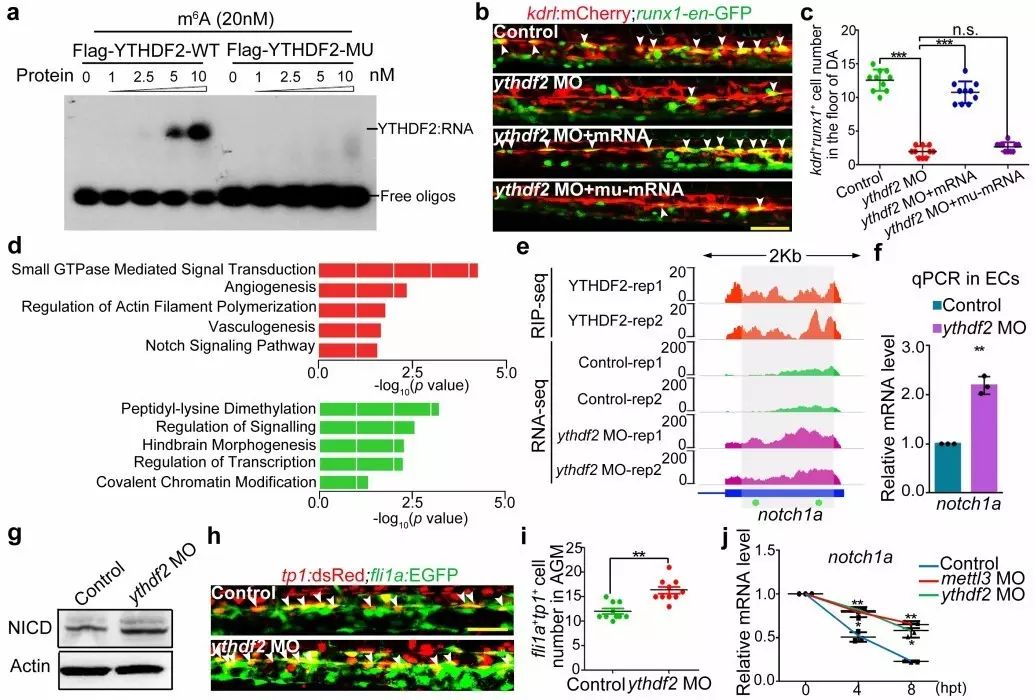
图4. YTHDF2介导的mRNA稳定性参与m
6
A对Notch信号的调控





