徐佳熹、孙媛媛、黄翰漾、
东楠、
刘鹭、黄昭宇、
杨希成、
李博康、
王楠、
王佳慧、
李昶霖、蔡莹琛(港)、李伟(港)
、周逸(港)
、余克清(港)
2021年5月10日至5月16日期间,CDE批准新药0款,FDA批准新药1款,具体情况如下;
2021-5-12批准上市Heron Therapeutics的布比卡因+美洛昔康注射剂,用于术后镇痛,获批类型为
Type 4 - New Combination,且获得优先审批(
PRIORITY)
资格。
国内IND化药21款,生物制品23款;
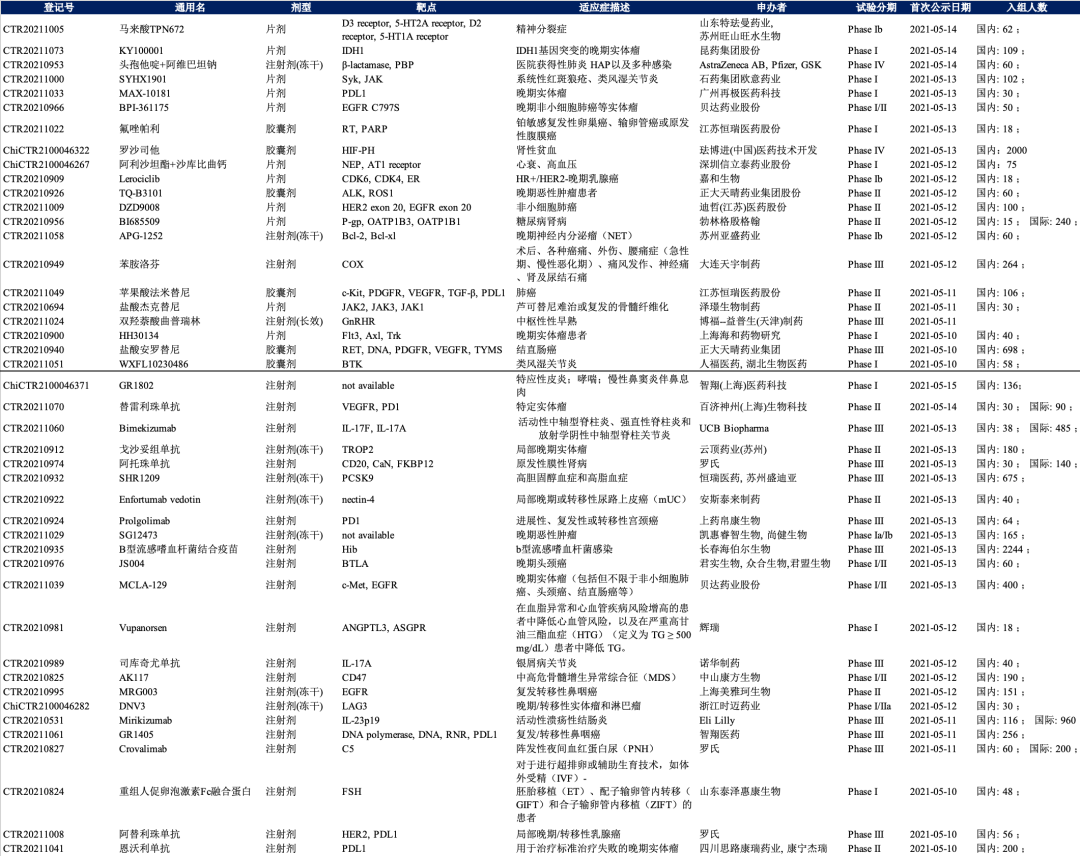
资料来源:FDA,CDE,医药魔方,兴业证券经济与金融研究院整理
1. Biogen 眼科疾病基因疗法 II/III 期临床研究失败
5月14日,Biogen宣布代号为XIRIUS的II/III期临床研究未达到主要临床终点。该研究旨在评估基因疗法cotoretigene toliparvovec(BIIB112)一次性治疗x-连锁视网膜色素变性(XLRP)患者的疗效。
XLRP是一种罕见的遗传性视网膜疾病,最常见的原因是合成功能性视网膜色素变性GTPase调节蛋白(RPGR)的基因发生突变,导致视网膜感光细胞退化,视网膜色素沉积并逐渐累积,最终导致失明。此疾病主要影响男性,大多数患者的最初症状是晚上看东西困难,接着是视野受限,到40岁时最终失明。
每10万人中大约有2-4名男性被诊断为XLRP,在基因突变类型已知的XLRP病例中,大约75%-90%由RPGR基因突变引起。此疾病目前没有获得批准的治疗方法。2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,视网膜色素变性被收录其中。
Cotoretigenetoliparvovec (BIIB112)是一种处于研究阶段的、基于AAV8载体的视网膜下注射基因疗法,该疗法旨在为XLRP患者提供全长功能性RPGR,通过替换该基因,可使患者RPGR蛋白水平增加,这可能会潜在地减缓、停止或防止 RPGR相关XLRP患者的光感受器的进一步退化。
XIRIUS研究是首个在人类患者中开展的多中心、随机、三臂剂量递增和剂量扩大研究,确诊为XLRP的男性接受视网膜下注射BIIB112治疗。Part I 部分是为期24个月的剂量递增研究(n=18,18岁),Part II 是为期12个月的剂量扩大研究(n=32,随机年龄≥10岁),根据益处/风险评估从Part I 中选择高剂量和低剂量,与安慰剂组进行比较,以评估疗效和安全性。治疗结束后,还会对Part I、Part II 中的受试者进行长达5年的随访研究。该研究结果显示,接受治疗患者和未接受治疗患者眼睛黄斑完整性评估(MAIA)微视野检查结果未显示出统计学意义的显著改善,未达到主要终点,但是在几个临床相关的预先设定的次要终点观察到阳性趋势。
2. 阿斯利康GLP-1/GCGR双重激动剂申报临床
5月14日,国家药监局官网显示,阿斯利康胰高血糖素样肽-1(GLP-1)和胰高血糖素受体(GCGR)双重激动剂Cotadutide注射液临床申请已获CDE受理。

资料来源:
CDE,兴业证券经济与金融研究院整理
Cotadutide是阿斯利康开发的first-in-class胰高血糖素样肽-1/胰高血糖素(GLP-1/GC)双重受体激动剂,正在被开发用于治疗2型糖尿病T2DM、肥胖、非酒精性脂肪肝、慢性肾病。
2020年8月,欧洲肝病学会(EASL)年会上,阿斯利康公布了cotadutide针对超重或肥胖的T2DM患者的一项IIb期临床研究数据。该研究入组了834例超重或肥胖的T2DM患者,旨在评估cotadutide的整体代谢效应,并对肝脏生物标志物进行了探索性分析。研究中,受试者随机接受为期54周的安慰剂、开放标签利拉鲁肽(1.8mg,每日1次)或皮下注射cotadutide(100μg,200μg,300μg,每日1次)治疗。
研究结果显示,在超重/肥胖T2DM患者中,cotadutide在200μg剂量下与GLP-1受体激动剂利拉鲁肽具有相似的减肥效果,300μg剂量下体重和ALT降低幅度更大。NFS和FIB-4的改善也非常令人鼓舞。数据支持了在NASH患者中对cotadutide进行前瞻性临床试验的必要性。
但是cotadutide(300μg)组高达40%的患者出现恶心、呕吐,而相应的利拉鲁肽组和安慰剂组分别为16%、10%。Cotadutide(200μg)组中近20%的患者出现呕吐,而相应的利拉鲁肽组和安慰剂组分别为3%、5%。
3. Biogen收购治疗急性缺血性卒中新药TMS-007
5月12日,Biogen宣布行使其选择权,从TMS株式会社收购了一种治疗急性缺血性卒中的研究药物TMS-007。
TMS-007是一种小分子纤溶酶原激活剂,其作用机制与打破血栓和潜在抑制血栓部位的局部炎症有关。这种独特的组合可以将TMS-007定位为急性缺血性卒中患者的下一代潜在溶栓药物,目的是与目前批准的溶栓药物相比,提供一个延长的治疗窗口。
Biogen收购TMS-007的决定是基于2a期研究的积极数据。这项随机、安慰剂对照、递增剂量2a期研究包括90名日本参与者(TMS-007 52例,安慰剂38例)。该研究的主要终点是通过国家卫生研究院卒中量表评估的恶化程度为4分以上症状性颅内出血(sICH)发生率和安全性。
该研究达到了其主要终点,TMS-007治疗组没有出现症状性颅内出血(sICH),接受安慰剂治疗的患者发生率为3%,证明了TMS-007对脑血管重新开放以及患者功能恢复的积极影响。患者在中风症状出现后12小时内服药:接受TMS-007的患者平均治疗时间为9.5小时,接受安慰剂的患者平均治疗时间为9.3小时。所有接受TMS-007治疗的患者的剂量都超过了批准的溶栓药物的时间窗。
此外,在90天时,TMS-007对次要终点功能独立性有显著改善,在接受TMS-007治疗的患者中,40%的患者在改良Rankin量表(一种日常生活独立性的衡量标准)上得分为0或1,表明没有残留症状或无明显残疾,而接受安慰剂治疗的患者中,这一比例为18%(P=0.05)。在接受TMS-007治疗的可见闭塞的患者中,血管再通的客观血管造影证据支持了这一点。通过磁共振血管造影测量,接受TMS-007治疗的患者再通率为58.3%(14/24),而接受安慰剂治疗的患者再通率为26.7%(4/15)(优势比4.23;95%CI:0.99~18.07)。
急性缺血性卒中是一种可能使人衰弱甚至致命的脑血管疾病,是世界范围内的第二大死亡原因,每年约有1300万例卒中病例,550万例死亡,急性缺血性卒中幸存者因大脑受到不可逆转的损伤而出现持续的功能缺陷。目前急性缺血性卒中仍有大量未得到满足的医疗需求,需要新的疗法,既能改善临床结果,提高疗效和安全性,又能延长中风患者接受溶栓治疗的时间。
由于已批准的溶栓药在以后的时间窗口中的利益风险状况,这些药物的使用受到限制。根据美国心脏协会的研究,sICH是目前溶栓疗法组织纤溶酶原激活剂(tPA)最令人害怕的并发症,它通过溶解阻止血液流向大脑的血凝块来发挥作用。在卒中发作后9小时的时间窗内,在对照研究中,接受tPA治疗的患者发生脑出血的比率高达6%。
作为收购TMS-007的一部分,Biogen将一次性支付1800万美元。如果TMS-007达到某些开发和销售额里程碑,TMS有资格获得额外的3.35亿美元里程金付款。TMS也有资格在全球年度净销售额中获得高个位数至低双位数百分比的分层版税。收购后,Biogen将全权负责TMS-007的开发、制造和商业化相关的成本和费用。Biogen目前正在评估TMS-007临床开发的下一步,包括全球研究计划。
4. 默沙东Keytruda早期三阴性乳腺癌新辅助/辅助III期研究达到EFS主要终点
5月14日,默沙东宣布代号为KEYNOTE-522的关键III期临床研究获得积极结果,达到了无事件生存(EFS)的第2个主要终点。根据独立数据监测委员会(DMC)的中期分析,与单纯术前化疗相比,Keytruda(帕博利珠单抗)联合化疗作为新辅助治疗,术后继续单药作为辅助治疗可使高危早期三阴性乳腺癌(TNBC)患者EFS具有统计学和临床意义的改善。Keytruda是首个在TNBC新辅助/辅助治疗中显示出EFS显著统计学意义的PD-1疗法。
该研究的另一个主要终点为pCR(病理完全缓解)的分析数据已公布于2019年欧洲医学肿瘤学会(ESMO)大会上,并发表在了《新英格兰医学杂志》上。研究结果显示,在早期TNBC患者中,无论PD-L1状态如何,与单纯化疗相比,Keytruda联合化疗作为新辅助治疗组患者的pCR的增加具有统计学意义。
此前,默沙东基于pCR数据和中期EFS研究结果,向FDA提交了Keytruda治疗高危早期TNBC患者的补充生物制剂许可申请(sBLA),然而,2021年3月,默沙东收到FDA完整回复函,FDA肿瘤药物咨询委员会以10:0的投票结果认为,是否批准此项适应症需在得到KEYNOTE-522研究更多数据后再做决定。如今,这项研究获得积极结果,预计该项适应症也能够顺利获得FDA批准。
KEYNOTE-522研究是一项III期随机双盲试验(NCT03036488),旨在评估Keytruda(抗-PD-1)联合化疗作为术前新辅助治疗,术后继续单药作为辅助治疗vs. 化疗作为新辅助,术后辅助安慰剂治疗高危早期三阴性乳腺癌(TNBC)患者的疗效。两个主要终点分别是pCR和EFS。该研究共招募了1174例患者,按2:1随机分配,分别接受以下两组方案治疗:
A) Keytruda (每3周1次)+紫杉醇(每周1次)+卡铂(每周或每3周1次)共4个周期,随后接受Keytruda +环磷酰胺+阿霉素或表柔比星(每3周1次)共4个周期作为术前新辅助治疗。术后接受9个周期的Keytruda (每3周1次)作为辅助治疗或;
B) 安慰剂(每3周1次)+紫杉醇(每周1次)+卡铂(每周或每3周1次)共4个周期,然后接受安慰剂+环磷酰胺+阿霉素或表阿霉素(每3周1次)共4个周期作为术前新辅助治疗。术后接受9个周期的安慰剂(每3周1次)作为辅助治疗。
5. 首款角膜塑形隐形眼镜获FDA批准上市,用于治疗儿童近视
5月12日,强生旗下医疗器械公司Johnson & Johnson Vision宣布,美国FDA已批准其开发的夜间近视治疗眼镜ACUVUE Abiliti上市。这是首个也是唯一一个获得FDA批准的用于治疗近视的角膜塑形(orthok)隐形眼镜。
Abiliti是一款优化的夜间视力矫正隐形眼镜,其所连接的创新型软件是一个复杂并且用户友好的工具,其能够提供角膜形状精确测量数据,并能够准确地指导专业眼科护理人员完成整个验配过程,能够达到大约90%的一致且成功的首次验配率。
Abiliti夜间角膜塑形隐形眼镜通过患者眼睛独特的角膜形状专门设计,能够临时重塑角膜,并且仅需在夜间佩戴。临床研究表明,其可作为治疗近视的一种安全、有效的方法,在2年的时间里,Abiliti夜间近视治疗眼镜可使近视儿童的眼轴伸长平均减少0.28毫米,眼轴伸长是导致近视的主要原因。
6. 诗健生物 Trop-2 ADC申报临床,国内第6家
5月13日,CDE官网显示,诗健生物/东曜药业重组人源化抗Trop2单抗-SN38偶联物临床试验申请已获国家药监局受理,该产品是国内第6家申报临床的Trop-2 ADC。

资料来源:
CDE,兴业证券经济与金融研究院整理
目前国内尚无该靶点ADC药物获批上市。研发进度最快的Trop-2 ADC是云顶新耀在2019年以8.35亿美元的交易从吉利德科学公司引进的Trodelvy(Sacituzumab Govitecan-Hziy,注射用戈沙妥组单抗),该款产品的上市申请已于昨日获得了CDE拟优先审评资格,用于治疗接受过至少两线既往治疗的转移性三阴性乳腺癌。
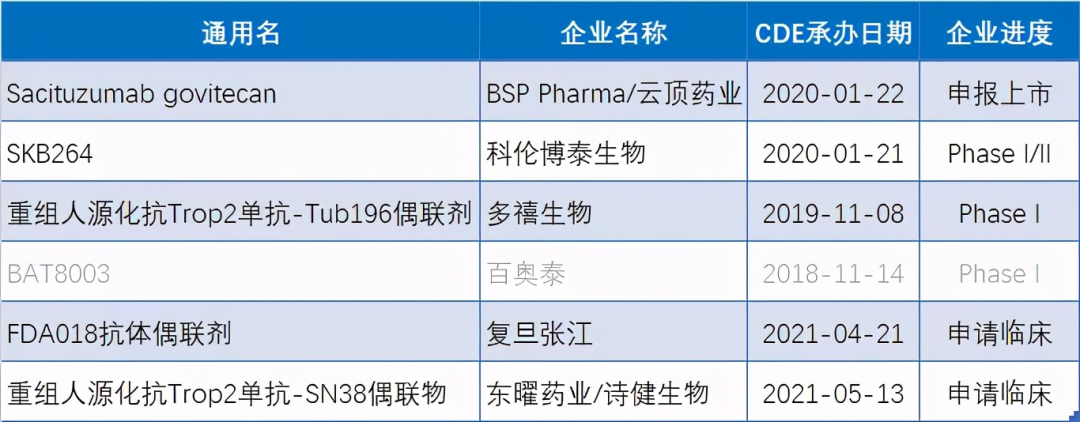
资料来源:
CDE,医药魔方,兴业证券经济与金融研究院整理
科伦博泰、多熙生物、复旦张江、百奥泰也已申报该靶点ADC。其中百奥泰已于今年3月5日终止了Trop-2 ADC 项目BAT8003的开发。

使用本研究报告的风险提示及法律声明
兴业证券股份有限公司经中国证券监督管理委员会批准,已具备证券投资咨询业务资格。
本报告仅供兴业证券股份有限公司(以下简称“本公司”)的客户使用,本公司不会因接收人收到本报告而视其为客户。本报告中的信息、意见等均仅供客户参考,不构成所述证券买卖的出价或征价邀请或要约。该等信息、意见并未考虑到获取本报告人员的具体投资目的、财务状况以及特定需求,在任何时候均不构成对任何人的个人推荐。客户应当对本报告中的信息和意见进行独立评估,并应同时考量各自的投资目的、财务状况和特定需求,必要时就法律、商业、财务、税收等方面咨询专家的意见。对依据或者使用本报告所造成的一切后果,本公司及/或其关联人员均不承担任何法律责任。本报告所载资料的来源被认为是可靠的,但本公司不保证其准确性或完整性,也不保证所包含的信息和建议不会发生任何变更。本公司并不对使用本报告所包含的材料产生的任何直接或间接损失或与此相关的其他任何损失承担任何责任。
本报告所载的资料、意见及推测仅反映本公司于发布本报告当日的判断,本报告所指的证券或投资标的的价格、价值及投资收入可升可跌,过往表现不应作为日后的表现依据;在不同时期,本公司可发出与本报告所载资料、意见及推测不一致的报告;本公司不保证本报告所含信息保持在最新状态。同时,本公司对本报告所含信息可在不发出通知的情形下做出修改,投资者应当自行关注相应的更新或修改。










