本文介绍了米测技术中心原创的一篇关于C–H官能化合成策略的研究。该研究旨在解决C–H官能化合成方法在应用中存在的问题,如选择性和立体化学控制问题。文章提出了一种新的合成策略,利用C–H官能化反应,成功合成了一种复杂天然产物(–)-柱孢环酚A。该策略涉及多个实验室合作,体现了多机构合作的成效。研究亮点在于展示了催化剂控制的不对称C–H功能化以及C–H功能化在全合成中的应用。
近年来,C–H官能化已成为一种可行的有机合成策略。然而,C–H官能化合成方法的应用存在选择性和立体化学控制问题。
C–H官能化合成方法在应用中存在选择性和立体化学控制问题。选择性C–H键官能团化和立体化学控制在合成具有挑战性的天然产物时非常重要。
研究提出了一种新的C–H官能化全合成策略,利用手性四羧酸二铑催化实现伯、仲位的C–H官能化,辅以钯催化的C (sp 2 )-C (sp 2 )交叉偶联,快速形成具有高区域、非对映和对映选择性的大环母核和所有立体中心。
该研究展示了催化剂控制的不对称C–H功能化,通过TPCP配体在Rh 2催化剂中对远端未活化的亚甲基位点进行不对称C–H官能化来形成C–C键的方法。此外,研究还实现了C–H功能化在全合成中的应用,特别是在构建[7.7]对环芳烷天然产物(–)-柱孢环酚A的过程中。
该研究成功合成了一种复杂天然产物(–)-柱孢环酚A,并通过C–H官能化生成六个C–C键和四个C–O键。这种新的合成策略涉及多种催化剂控制的对映选择性和非对映选择性C–H功能化。

特别说明:
本文由米测技术中心原创撰写,旨在分享相关科研知识。因学识有限,难免有所疏漏和错误,请读者批判性阅读,也恳请大方之家批评指正。
原创丨
米测MeLab
编辑丨
风云
研究背景
近年来,C–H 官能化已成为一种越来越可行和易于实施的有机合成策略。目前已经开发出许多高效的选择性C–H官能化方法,如使用导向基团、自由基转化和催化剂控制的基团转移反应。
关键问题
然而,C-H官能化合成方法的应用主要存在以下问题:
1、C-H官能团化方法的应用存在选择性问题
在C-H键官能团化中,实现位点选择性氢原子转移通常依赖于底物控制,底物上具有不同键解离能、极性、空间位阻或电性的C-H键决定了位点选择性。然而,有机分子中通常包含多个具有类似性质的sp
3
C-H键,给选择性C-H键官能团化带来了巨大的挑战。
2、C-H官能团化方法存在立体化学控制问题
在构建具有挑战性的天然产物时,C-H功能化需要高区域、非对映和对映选择性,这要求专门的过渡金属催化系统具有高度的立体电子精度。这种立体选择性的控制在实际操作中可能存在困难。
新思路
有鉴于此,
埃默里大学Huw M. L. Davies、加州理工大学Brian M. Stoltz等人
报道了一种(-)-柱孢环酚A的合成策略,
使用10个C-H官能化反应,以高对映选择性和高效率(17步)得到了一条精简的路线
。利用手性四羧酸二铑催化实现了伯、仲位的C-H官能化,并辅之以钯催化的C (sp
2
)-C (sp
2
)交叉偶联,快速形成了具有高区域、非对映和对映选择性的大环母核和所有立体中心。使用后段钯催化的四重C (sp
2
)-H乙酰氧基化反应来安装双间苯二酚部分。这项研究体现了多实验室合作如何对复杂的全合成工作实现实质性的现代化。
技术方案:
1、提出了C-H官能化全合成策略
作者通过C-H官能化策略,从双酰胺
2
合成(–)-柱孢环酚A,引入间苯二酚官能基,利用铑催化构建C-C键和大环,制备芳基卤化物
6
。
2、展示了不对称C–H功能化及直接环化二聚
作者利用Rh
2
催化剂对芳基重氮乙酸酯
7
和反式-2-己烯
8
进行一级C-H官能化,高效合成大环
15
,构建了[7.7]对环芳烃核心及四个立体中心。
3、开发了逐步大环化并实现了全合成
作者开发了一种逐步合成大环
15
的高效方法,通过Rh
2
催化剂处理构建立体二联体,最终合成了(–)- 柱孢环酚A。
技术优势:
1、展示了催化剂控制的不对称C-H功能化
作者在研究中展示了通过使用TPCP配体在Rh
2
(2-Cl-5-Br)TPCP
4
催化剂中对远端未活化的亚甲基位点进行不对称C-H官能化来形成C-C键的方法,实现了以高区域、非对映和对映选择性构建具有挑战性的键。
2、实现了C-H功能化在全合成中的应用
本工作强调了C-H功能化在全合成中的应用,特别是在构建[7.7]对环芳烷天然产物(–)-柱孢环酚A的过程中,通过10个C–H官能化生成的六个C–C键和四个C–O键,这些键具有较高的位点选择性和立体选择性。
技术细节
合成方案
在对(–)- 柱孢环酚A的逆合成分析中,作者提出了一种通过C-H官能化实现快速分子简化的策略。作者设想从双酰胺
2
出发,通过亲核试剂添加合成
1
,并计划在后期通过四重Weinreb酰胺定向的C-H乙酰氧基化引入2,6-间苯二酚官能基。铑(II)催化的二级C(sp
3
)-H官能化将用于构建C-C键和大环化,而芳基卤化物
6
的制备则依赖于对映选择性铑(II)催化的一级C-H官能化。这一策略旨在揭示重氮酯
5
,为合成等价己烷提供新途径。
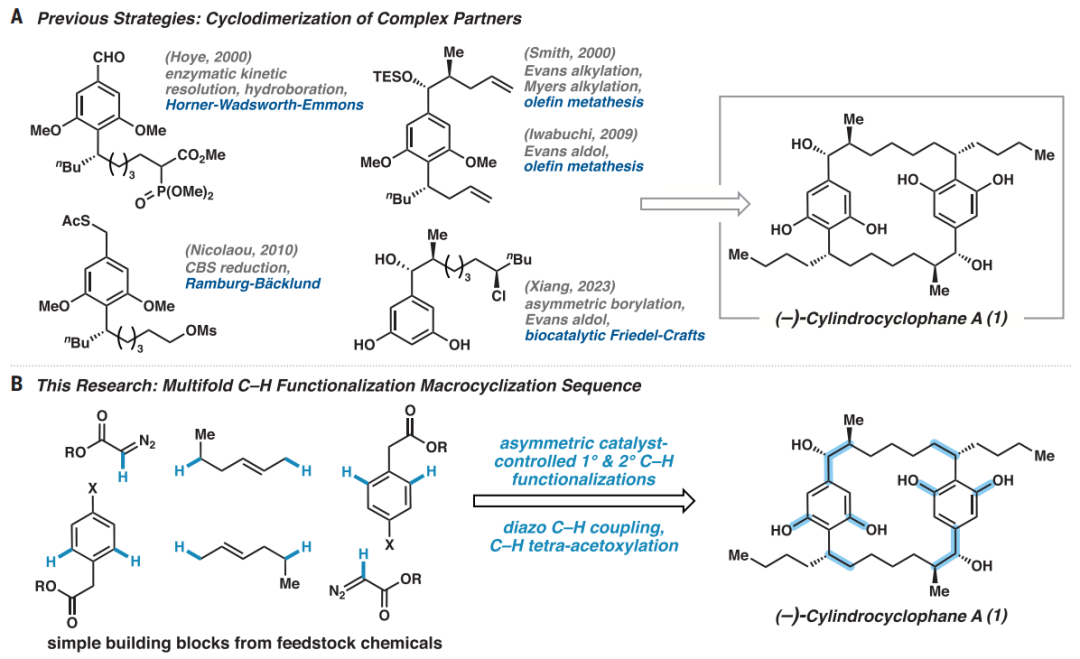
图 针对(–)- 柱孢环酚A的策略

图 (−)-柱孢环酚A的逆合成
不对称C–H功能化及直接环化二聚
作者对芳基重氮乙酸酯
7
和反式-2-己烯
8
进行了选择性一级C-H官能化,使用空间位阻Rh
2
(R-p-PhTPCP)
4
催化剂,实现了高区域和对映选择性,得到产物
9
(73%产率,96% ee)。随后,通过Crabtree催化剂氢化得到碘化物
10
。接着,通过钯催化C(sp
2
)-C(sp
2
)交叉偶联合成芳基重氮乙酸酯
12
。在构建[7.7]对环芳烃骨架的过程中,发现Rh
2
(R-2-Cl-5-BrTPCP)处理
12
可生成大环
15
,产率中等(19%,非对映体比6:1)。X射线衍射分析确认了
15
的绝对和相对构型,该一锅反应在一个步骤中组装了天然产物的[7,7]对环芳烃核心及四个新立体中心,尽管重氮乙酸酯
12
的直接环二聚化存在材料通量和纯化挑战。

图 [7.7]对环芳烷骨架的合成
逐步大环化及全合成
为了提高材料通量,作者开发了一种逐步合成大环
15
的方法。使用Rh
2
(R-2-Cl-5-BrTPCP)
4
催化剂处理重氮乙酸酯
12
,高效生成芳基碘化物
13
,具有高立体和区域控制性(68%产率,19:1 dr)。随后,通过钯催化交叉偶联和Rh
2
催化的大环化,构建了大环
15
的第二个立体二联体(产率70%)。这种方法允许一次性制备超过1.2 mmol的大环
15
,并通过重结晶分离为单一非对映体。通过直接环二聚化或分步大环化从芳基重氮乙酸酯
7
合成大环
15
,高效构建大环核心和六个立体中心。大环
15
的三氯乙酯转化为双Weinreb酰胺
16
,剩余三氟乙酯水解得到双羧酸
17
,进一步转化为双苄基乙酸酯
3
。最后,通过四-C(sp
2
)-H乙酰氧基化生成大环
2
,完成所有C−H官能化步骤。安装两个丙基侧链的尝试中,发现将苯酚乙酸酯转化为甲醚并进行还原裂解、Wittig烯化和氢化,最终得到(–)- 柱孢环酚A (
1
)。
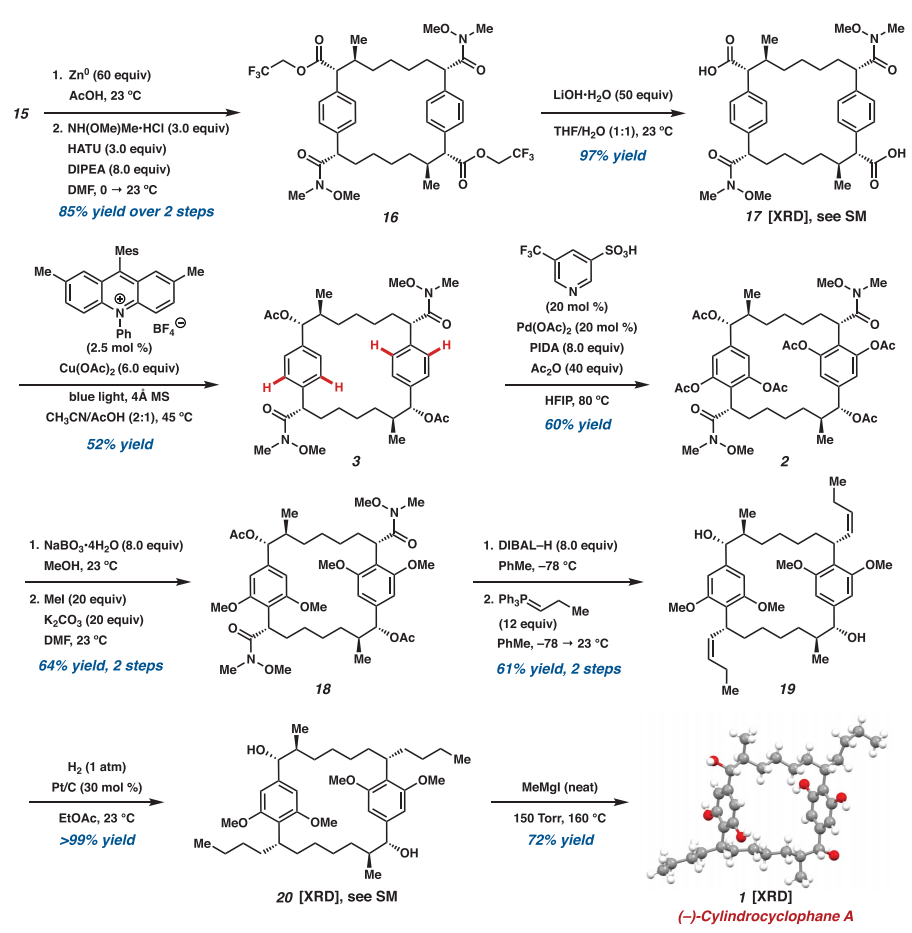
图 最终C–H官能化为 (–)- 柱孢环酚A
展望
总之,本工作的合成涉及较短的17步或更具可扩展性的19步序列,从商业起始材料开始,使用10个C–H功能化反应形成六个C–C键和四个C–O键。具体来说,该路线展示了四种催化剂控制的对映选择性和非对映选择性C–H功能化,以生成天然产物的所有六个立体中心,以及两种钯催化的重氮羰基化合物 C-H功能化和四种酰胺定向C-H乙酰氧基化。这项研究体现了多机构合作和C–H 功能化的强大功能,这是一种将低成本材料选择性转化为高度功能化和立体化学复杂构件的有利技术。
参考文献:
AARON T. BOSSE, et al. Total synthesis of (−)-cylindrocyclophane A facilitated by C−H functionalization. Science, 2024, 386(6722): 641-646.
DOI: 10.1126/science.adp2425
https://www.science.org/doi/10.1126/science.adp2425
纳米人学术QQ交流群
(
加群方式:请
备注:姓名-单位-研究方向(无
备注请恕不通过),由管理员审核后入群。)
一维材料群-1:463352826
二维材料群-2:1075914107
复合材料群-2:834770793
可穿戴器件学术群:1032109706
光催化群-2:927909706
电池讨论群-1:418038617





