知了网祝广大高考考生金榜题名
文章转载需获得知了网同意,并注明作者、来源、原文出处等。
在专利这个圈儿里,我们周围不乏有生物专业出身的代理人,当看着他们与发明人讨论技术方案时,随口脱出一堆高大上的专业术语,如微卫星分子标记、高保真Taq酶、遗传多态性,外加复杂的实验步骤,利用DNAMAN软件寻找亲缘关系较近物种的保守序列,根据保守序列设计引物,通过PCR扩增、毛细管电泳筛选多态性引物……小编不禁感叹生命科学真是复杂,因为生命本身就很复杂。
生命的全部信息都记录在基因里,基因控制着生物体的性状,有时还会与某些疾病的发生发展有关。随着人类基因组测序计划的完成,人们对数以亿计的ATGC序列中包涵着什么信息产生了浓厚的兴趣。这些信息是怎样控制生物体发育的?基因组本身又是怎样进化的?毫无疑问,我们已经开始从积累生物数据逐渐向解释这些数据转变,生物信息学正是在这一的背景下产生的交叉学科。
生物信息学以核酸序列、氨基酸序列为研究对象,本着相似序列在结构和功能上也会有一定程度的相似的规律,对蛋白质的结构和功能进行预测,进而再进行其他深入的研究,如药物设计等。利用生物信息学进行科学研究的出发点是理论推测,从理论推测出发,然后再回到实验中去,追踪或验证这些理论假设。目前,全部基因都可通过测序知晓其序列,并以计算机可读的方式存储在数据库中,这种研究方法对于每14个月体量就会翻一倍的基因数据来说,显然更高效。
序列比对是生物信息学的基础,通过将新测定的序列与数据库中已知的序列进行比对,可以查找到相似序列,发现与结构和功能相关的保守序列片段。我们是如何进行比对的呢,简单举一个例子:新测定序列(S1)=GCCCTAGCG,已知序列(S2)= GCGCAATG,在不考虑空位的情况下,S1和S2有以下两种比对方式:
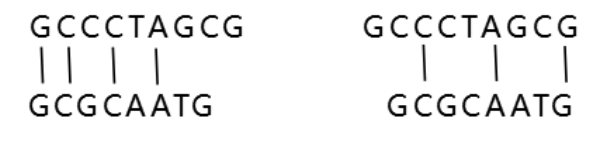
序列比对查找的是最大匹配序列,我们设定匹配得1分,失配得0分,那么上述两种比对方式的得分分别为4、3,优选第1种比对方式。接下来S1还要和数据库里的S3、S4、S5…Sn进行比对,分值最高的,为最相似的序列。
但实际上远不止这么简单,一是物种在进化过程中会发生基因突变,出现碱基置换、缺失或插入现象,因此,我们在寻找最优比时需要考虑删除或插入空位,理论上讲缺失或插入碱基的现象发生频率较低,所以在引进空位时要进行罚分;二是不同碱基或氨基酸的置换,最终产生影响程度不同,特别是对于一些氨基酸,可以很容易相互取代,而不改变蛋白质的理化性质,因此,我们又设计了在不同情况下应用的多种得分矩阵,如颠换矩阵、疏水矩阵、PAM和BLOSUM等。此外,随着比对序列长度的增长,很难列举出所有可能的比对,然后计算得分,这时候寻找最优比对则需要借助算法和计算机编程来实现。
目前,针对两两序列比对的算法也有:基于动态规划思想的Needleman-Wunsch算法和Smith-Waterman算法,基于启发式思想的Blast算法等。动态规划算法,是将两条序列分别作为矩阵的两维进行打分,这样序列会被分解成更短的小序列进行比对,以小序列的最优比对找原序列的最优比对。例如,我们利用Needleman- Wunsch算法对S1和S2进行全局比对,S1横向展开,S2纵向展开,匹配记1分,不匹配扣1分,空位扣2分,得到如下得分矩阵:
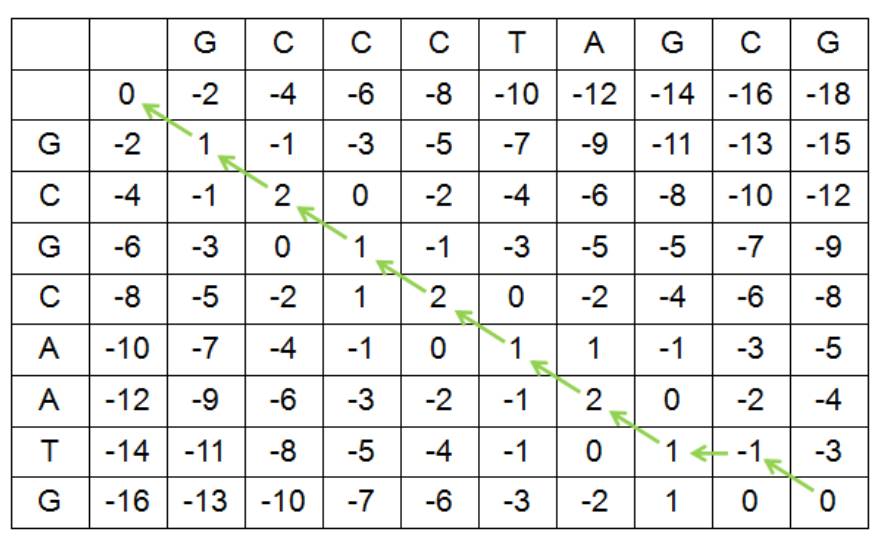
通过回溯可以在得分矩阵里找到最优比对路径。如果表格里的得分在横纵方向上对应的S1和S2的碱基相同,则回溯到左上方单元格,如果不同,则回溯到左上方、上方、左边最大数值的单元格,最大数值相同时,按照左上方、上方、左边的顺序进行回溯。从右下角回溯至左上角的路径为S1和S2的最优比对路径。根据回溯路径可以写出比对方式,回溯到左上方单元格的,该单元格对应的碱基分别为S1和S2对比时对应的碱基,回溯到上方单元格的,S1以空位对应S2的碱基,回溯到左边单元格的,S2以空位对应S1的碱基。因此,下面所示的比对就是S1和S2的最优全局比对:
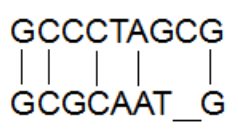
如果是局部比对,找出某一片段的最优比,则需要使用Smith-Waterman算法,在同样的得分函数下,两个0字符得分为0,得分为负数的记做0,S1和S2局部比对可以得到如下得分矩阵:
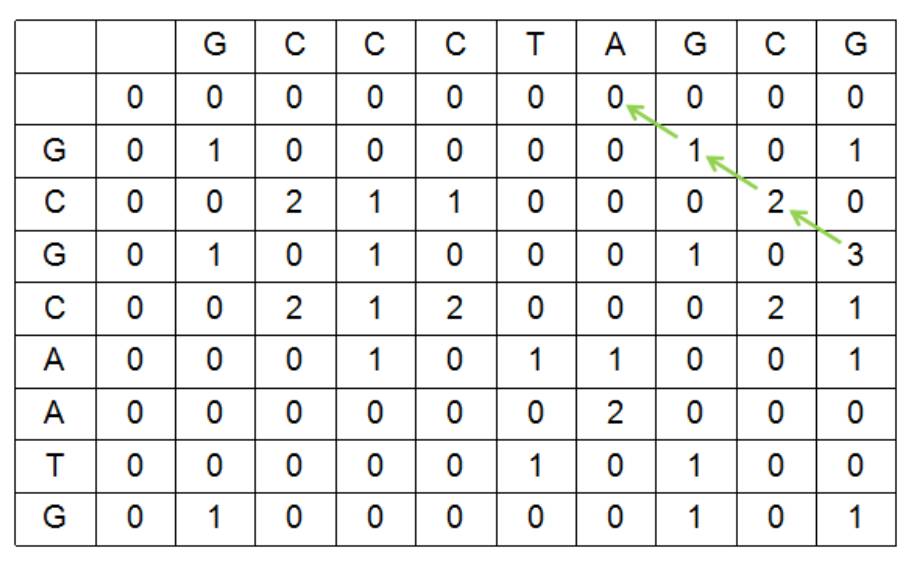
局部比对从得分最高的单元格进行回溯即可,根据绿色箭头指示的回溯路径,S1和S2的最优局部比对如下:
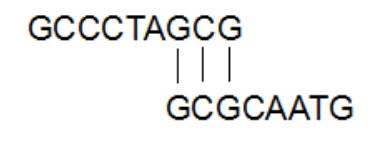
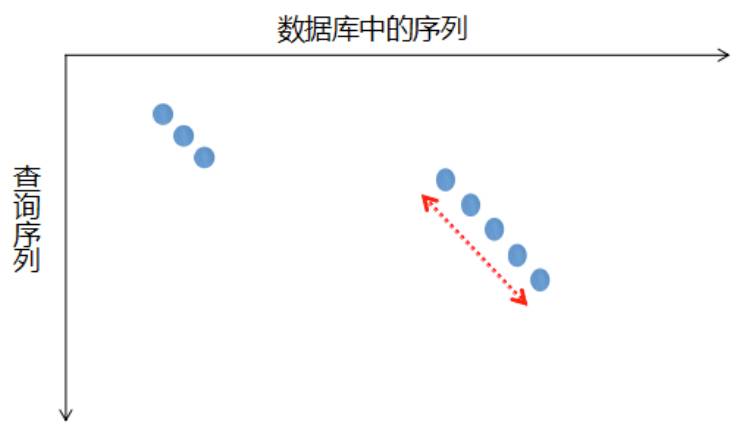
通过上面的例子我们发现,最优全局比对出现在主对角线区域,最优局部比对出现在与对角线平行的区域,如果有一种方法能够在这些区域里寻找最优比对,是不是就可以将矩阵简化,加快比对速度呢?Blast算法成功解决了这一问题。Blast算法首先将查询序列按顺序划分为长度为w的子序列,进行数据库搜索,找到匹配得分高于阈值的子序列对,也就是所谓的种子,而后以此为基础优先在对角线方向上延伸,找到高分片段对。Blast算法得到的结果会进行统计显著性分析,用期望值(expect value)表示,期望值E是指在随机的情况下,其它序列与查询序列相似度要大于这条显示的序列的可能性,所以它的分值越低越好。
基于Blast算法,DI Inspiro开发了生物序列检索,用户可根据核酸序列或或氨基酸序列进行相关专利的检索,系统收录了中国、奥地利、澳大利亚等17个国家和地区的3000余万条生物序列数据。对于核酸序列检索,我们可以输入检索序列,选择数据库以及算法。megablast适用于检索高度相似的序列,非连续的megablast会忽略一些错配,主要用于跨物种之间的序列比对,blastn适用于短序列检索,短到7个碱基的序列。
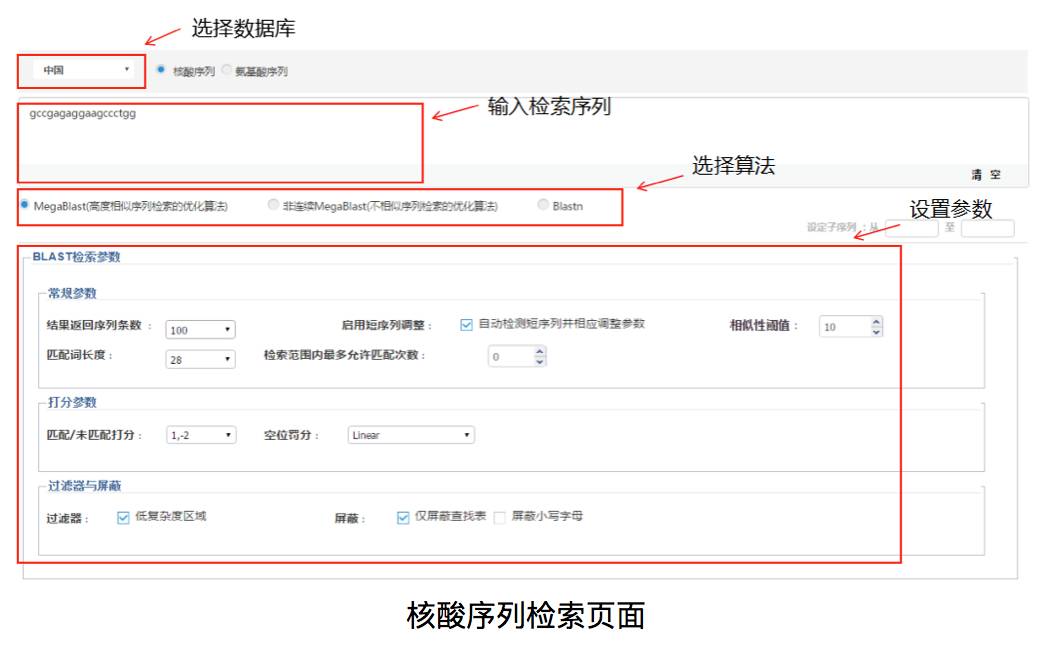
Blast检索参数一般设置为默认值,相信了解了前面的基础知识后,不难理解这些参数的含义:
结果返回序列条数是检索后显示的检索结果数量;Blast算法在确定种子前,需要将查询序列划分为w长度的小序列,w就是匹配词长度;启动短序列调整是指自动调整匹配词长度和其他参数以缩短检索时间;相似性阈值为期望值E,用于评价检索得到的某段序列与检索序列之间相似这一检索结果是否可靠,E值越低越好;检索范围内最多允许匹配次数用于限制与检索序列匹配的数量;
匹配/未匹配打分用于确定得分函数;空位罚分分为线性空位罚分和仿射空位罚分,线性空位罚分的计算公式是空位数与罚分相乘,仿射空位罚分将空位的长度也考虑在内,第一个空位罚分g,空位延伸罚分r;
过滤是将一些串联重复序列和低复杂度序列屏蔽掉,因为这两种区域可能会让BLAST找出一些虽然分数够高,但是其实和检索序列并不相关的序列。
我们输入一段序列gccgagaggaagccctgg,选择megablast算法,在默认参数下,执行检索,检索结果如下图所示。
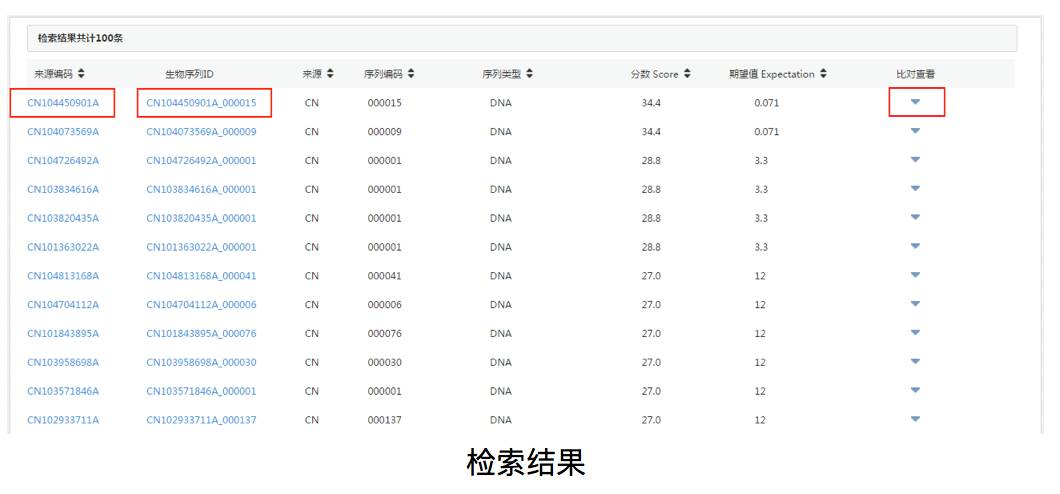
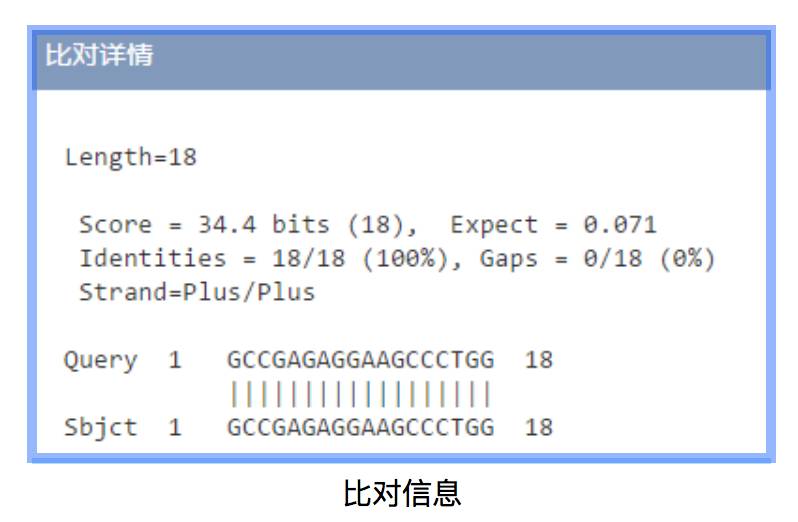
生物序列的检索结果按照分值由高到低排列,点击专利公布号、生物序列ID编码、比对查看可分别获取序列来源专利、序列生物信息、比对信息。
动态编程和基因序列比对,Paul Reiners
生物序列比对,黄宁
BLAST介绍,https://blast.ncbi.nlm.nih.gov/Blast.cgi
广宣时段:机构数据、企业数据、知识产权大数据,知了网已然一站式了,还需要寻找吗?

www.zhiqicha.com
注册用户参与活动
几乎百分之百中奖率
给你1个月VIP福利
和7天尝鲜体验
扫描二维码来参加吧
使用兑奖码+已注册知企查的手机号可获得哦

进入公众号,点击右下角也可砸金蛋
活动截止日期:6月9日
登陆知企查(http://www.zhiqicha.com/)
提供建议或意见,获取免费使用账号
关注知了网,参与更多活动。


想不想
读我们,知资讯,看世界
滑至底部点 阅读原文
知了网
知识产权云服务平台
找代理、搜专利、创意描述、自助撰写
www.izhiliao.com.cn 











