
BUSCO目前已经更新到第4版,之前只用来评估基因组为目标,现在还能够预测基因和做系统基因组学分析。
软件安装
官方提供了docker,conda和GitLab这三种方法,这里我只介绍conda。
conda create -n busco4 -c bioconda -c conda-forge busco=4.0.5
conda activate busco4
由于软件运行的时候会用到AUGUSTUS,AUGUSTUS运行的时候需要你额外设定2个环境变量,
AUGUSTUS_CONFIG_PATH
和
BUSCO_CONFIG_FILE
, 通过conda安装的这两个配置文件都在
/path/
to
/
miniconda3
/
envs
/
busco4
/
config
目录下,所以需要在
.
bashrc
或
.
zshrc
中加入下面这一行
export AUGUSTUS_CONFIG_PATH="/path/to/miniconda3/envs/busco4/config
export BUSCO_CONFIG_FILE="/path/to/miniconda3/envs/busco4//config/config.ini
这里的
/path/
to
/
miniconda3
指的是的conda安装路径,请按照实际情况替换。
软件运行
BUSCO4能够自动下载数据集,并进行分析。但是考虑到国内的网络环境,我建议直接去https://busco-data.ezlab.org/v4/data/lineages/ 下载自己所需的物种,比如被子植物可以下载
wget https://busco-data.ezlab.org/v4/data/lineages/embryophyta_odb10.2019-11-20.tar.gz
tar xf embryophyta_odb10.2019-11-20.tar.gz
后续假如你组装的基因组为
genome
.
fa
,
embryophyta_odb10
在
/data/
database
/
目录下,那么运行命令如下
busco -m geno -i genome.fa -l /data/database/embryophyta_odb10 -o busco4 -c 20 --offline &
和之前一样,运行结束后会有一个总体统计结果,这个文件在输出目录下,以
short_summary
.
作为前缀。
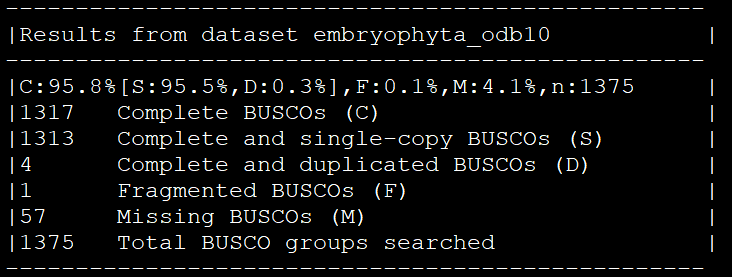
除此之外,这次BUSCO最大的升级在于,它不再只输出单拷贝基因,还会输出多拷贝基因。比如说,以我提供的参数进行运行,那么可以在
busco4
/
run_embryophyta_odb10
/
busco_sequences
下看到输出的三个目录
-
不完整序列: fragmented
busco
sequences
-
多拷贝序列: multi
copy
busco_sequences
-
单拷贝序列: single
copy
busco_sequences
此外,新版的还提供了一个
generate_plot
.
py
来展示结果, 你可以将多个物种的运行结果放在一个目录下,那么就能比较多个物种的情况
generate_plot.py -wd busco4

最后,如果你要使用BUSCO的话,建议直接用BUSCO 4即可,不需要再考虑之前的版本了。
参考资料: https://busco.ezlab.org/busco_userguide.html





