本期内容主要涉及 BWA + GATK 经典流程中的 GATK 部分。
FixMateInformation 步骤放在哪里比较合理?
上期的博文出来后, QQ 昵称为 Dreamooc 的朋友和我讨论了 FixMateInformation 这一步是否需要,以及放在流程中的哪一步比较合理的问题。仔细地翻阅了一些博文后,把最新的想法归纳了一下,供大家参考。
最初加上这一步分析是参考 Samtools 官网 (http://www.htslib.org/workflow/) 给出的流程。它里面提到 BWA 有时会产生不正常的 flag 信息,在进行其它分析前最好先整理 reads 的 mate 信息以及 flags。
SortSam 并没有依据 Mate 信息进行过滤,MarkDuplicates 如果仅仅是标记重复而不移除重复时,不会对 mate 信息产生影响,当时觉得 FixMateInformation 只要在用 GATK 分析之前运行了就可以 (GATK 默认会依据 mate 信息对数据进行过滤)。
如果 MarkDuplicates 这步把重复去掉,则会对 mate 信息产生影响,可以考虑在这步之后加上 FixMateInformation。
Dreamooc 提到有人在 IndelRealigner 之后添加了 FixMateInformation 这个步骤,因为局部重排会对部分reads 的 mate 信息产生影响。我查阅 GATK Forum 后,发现这个问题已经被 GATK 团队 解决了,详见:https://gatkforums.broadinstitute.org/gatk/discussion/5871/problem-with-indelrealigner-assigning-incorrect-mate-information-gatk-v3-4-46-gbc02625,以及 https://gatkforums.broadinstitute.org/gatk/discussion/1562/need-to-run-a-step-with-fixmateinformation-after-realignment-step
参考基因组质量很好的模式物种,可以查阅更多的信息,多尝试、多验证,以决定这一步是否需要以及放到哪里比较合适;如果是非模式物种,虽然基因组质量带来的影响远比这些步骤本身的影响大,但也要尽量做到最好,可以考虑在 pipeline 中把 FixMateInformation 这个步骤放在 SortSam 之前。
Indel realignment
对 mapping 得到的 bam 文件做完 Fix Mate Information、Sort 和 mark duplicates 处理后, 就可以进入 GATK 流程了。首先需要做 Indel realignment,其分为两步:第一步是 RealignerTargetCreator,第二步是 IndelRealigner。
ref=/your/path/to/ref.fa
GATK=/your/path/to/GenomeAnalysisTK.jar
java -jar "$GATK" -T RealignerTargetCreator -R "$ref" -I ${name}.dedup.bam -o ${name}.dedup.intervals --validation_strictness SILENT -nt 12
java -jar "$GATK" -T IndelRealigner -R "$ref" -I ${name}.dedup.bam -o ${name}.realigned.bam -targetIntervals ${name}.dedup.intervals --validation_strictness SILENT --filter_bases_not_stored
GATK 文档里面提到:如果用 HaplotypeCaller 或 MuTect2 等进行局部重新组装的 caller,可以移除 Indel realignment 这个步骤。但建议如果计算资源充裕的话,这个步骤是必要的,主要原因有以下两点:
省略这个步骤与否,得到的 SNPs 和 Indel 数目会有少许的差异;
在群体遗传学研究中,不是所有的分析都是基于基因型的,例如:比较常用的 ANGSD 是以 bam 为输入文件的,做 Indel realignment 可以提高分析结果的准确性;
对于人类或者其它模式物种来说,这个步骤还有一个其它的选项:--known indels.vcf,对文件内列出的 Indel 位点, 软件默认不对其进行操作。
GATK 默认会对数据进行过滤,比如 RealignerTargetCreator 的过滤情况为:
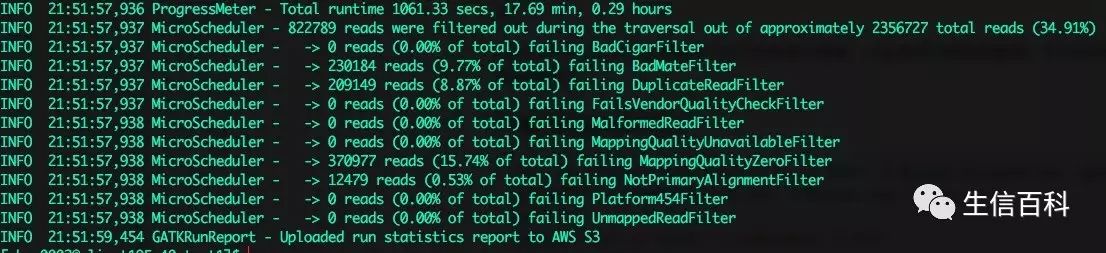
这些默认的过滤可以从每个工具的说明文档中查阅。
Recalibrate Bases
这个步骤对于非模式物种,以及缺少已知的、经过验证的 SNPs 和 Indels 的模式物种来说,不容易实现。我在目前的工作中没有进行这块内容的分析。
其它博客提到,可以同时运行 BWA + Samtools + Bcftoos 以及 BWA + GATK (无 Recalibrate Bases 步骤) 流程,经过严格的过滤后取两个结果的交集,以得到比较可靠的 SNPs 和 Indels 信息,用他们来做 Recalibrate Bases 的输入文件 (knownSites)。 我尝试过这个方法,在我的工作中效果并不理想。大家可以尝试一下,这样操作是否有效。
BaseRecalibrator 是用机器学习的方式来建立模型,以此来修正碱基质量值,可以很好的提高结果的准确性,值得大家尝试。
HaplotypeCaller
java -jar "$GATK" -T HaplotypeCaller -R "$ref" -I ${name}.realigned.bam --genotyping_mode DISCOVERY --emitRefConfidence GVCF -o ${name}.snps.indels.g.vcf
gvcfs=$(ls *.g.vcf)
combine="java -jar $GATK -T CombineGVCFs -R $ref"
for i in $gvcfs; do
combine+=" -V $i"
done
combine+=" -o Combined.g.vcf"
eval $combine
java -jar $GATK -T GenotypeGVCFs -R $ref -V Combined.g.vcf -o all.snp.indel.vcf
由于我并没有做 Recalibrate Bases,故用 Indel realignment 得到的文件作为输入文件。
HaplotypeCaller 有很多优点,在 GATK 的官网有介绍,大家可以去浏览。简单的说,它可以对 mapping 结果进行局部重新组装,已得到更加可靠的变异信息。在用 gvcf 模式时,HaplotypeCaller 会对每个个体生成一个中间文件 (gvcf,其记录这个个体的变异信息),将这些文件合并 (CombineGVCFs) 以后,可以用 GenotypeGVCFs 对多个样本进行 joint genotyping,以得到更准确的基因型。
我用 for 循环把所有的 gvcf 文件添加到参数中,这样更加简便、快捷。
如果样品较多,在合并 gvcf 文件时可以考虑合并成几个文件,把它们同时作为 GenotypeGVCFs 的输入文件。
相关阅读:
重测序分析流程及分析经验介绍