第一作者:周静,赫丙玲,黄璞
通讯作者:董玉明,王耀,黄宏文,章佳威
通讯单位:江南大学
论文DOI:10.1002/ange.202418459
将二氧化碳高效电还原(CO
2
RR)为乙醇,为同时生产增值化学品与利用可再生能源带来了希望。然而,由于在热力学上乙烯的生产更具优势,两者生产竞争激烈,使得该过程仍然面临挑战。在此项研究中,我们采用了一种创新方法,将全氟辛烷植入铜位点,从而构建了界面非共价相互作用,成功实现了二氧化碳还原产物偏好从乙烯到乙醇的显著逆转,乙醇与乙烯的比例达到约5.4。其中,1.83%F-Cu
2
O OIHs表现卓越,其乙醇FE高达约55.2%,乙醇分电流密度更是令人惊叹,达到166 mA cm
-2
,且在连续运行60小时后依然保持出色的稳定性。通过原位光谱以及DFT 计算等方法,揭示了C
8
F
18
增强界面氢键网络,促进活性氢物种生成,还优先推动了*CHCOH加氢生成*CHCHOH,从而提高乙醇选择性的机制,本研究凸显了利用非共价相互作用调控二氧化碳电还原制乙醇选择性的巨大潜力,为其更接近实际应用需求提供了重要机遇。
将过量的二氧化碳通过电还原反应转化为增值原料,是一种有前景的策略,能够应对环境和能源问题。在CO
2
RR的产物中,乙醇因其在能源密度(26.8 MJ/kg)、长期储存、可扩展性和运输性等方面的优势而备受关注。然而,乙醇和乙烯这两种主要的多碳(C
2+
)CO
2
RR 产物,它们分支于关键中间体*CHCOH,乙烯的形成在热力学上比乙醇更有利,这导致乙醇的选择性较差,通常比乙烯的选择性低2-3倍。因此,调控 CO
2
RR动力学以提高乙醇的选择性成为研究热点。
1. 本工作运用主客体策略,以SDS为软模板将F植入Cu
2
O纳米立方体,调控C
8
F
18
初始用量控制其含量,制备出C
8
F
18
-Cu
₂
O OIHs催化剂加速CO
2
电还原制乙醇。
2. 1.83%F-Cu
2
O OIHs催化剂增强氢键网络,稳定乙醇中间体,在流动池测试中乙醇FE达55.2%,乙醇/乙烯比约5.4,实现产物偏好逆转。
3. 本文利用原位光谱证明C
8
F
18
植入促进活性氢生成及*CHCOH加氢为*CHCHOH,并利用DFT计算印证F降低H
2
O解离能垒、增加*H覆盖度、改变反应路径能量,使反应向生成乙醇方向进行,阐释催化剂性能提升机制。
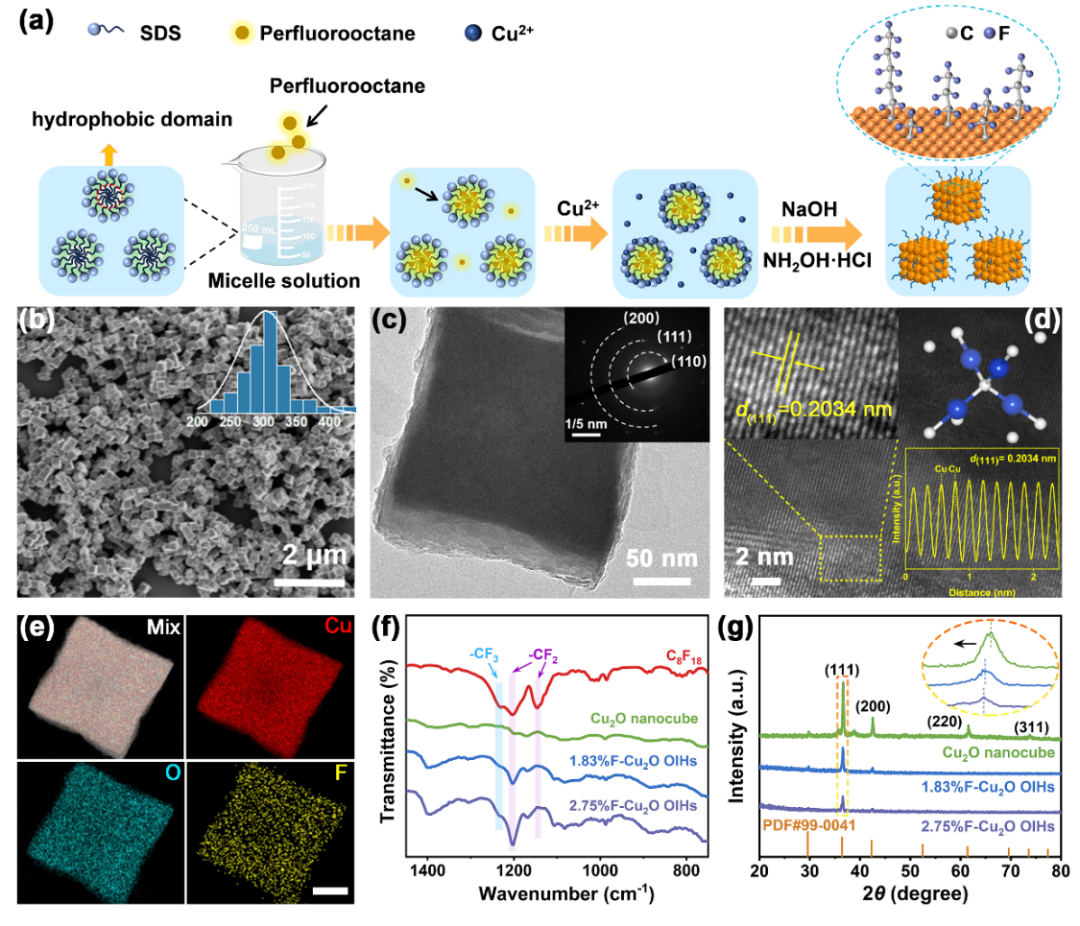
图 1:Cu
2
O 纳米立方体与x%F-Cu
2
O OIHs结构表征。(a) x%F-Cu
2
O
OIHs合成示意图;1.83% F-Cu
2
O OIHs的(b) SEM及直径分布图;(c) TEM及对应的SAED;(d) HRTEM及Cu
2
O晶胞;(e) EDS;三种催化剂的(f) FT-IR谱图;(g) XRD。
本文利用软模板法合成x%F-Cu
2
O OIHs,利用亲憎水性及静电作用形成装载C
8
F
18
内核且外聚集Cu
2+
的胶束,在还原剂和NaOH作用下引发Cu
2
O成核,同时C
8
F
18
被释放并植入。为了对合成的材料进行表征,进行了一系列测试。
一方面,SEM和TEM证明,
C
8
F
18
的植入并不影响Cu
2
O纳米立方体的形貌。SAED、HRTEM及XRD显示出Cu
2
O的晶型状态且F的植入导致了晶格膨胀。与此同时,EDS元素映射证实F在整个纳米立方体中均匀分布,此外,FT-IR光谱中的特征峰也证实了
C
8
F
18
的存在,通过这些测试,足以充分证明
C
8
F
18
成功植入Cu
2
O结构并对其产生了相应影响。
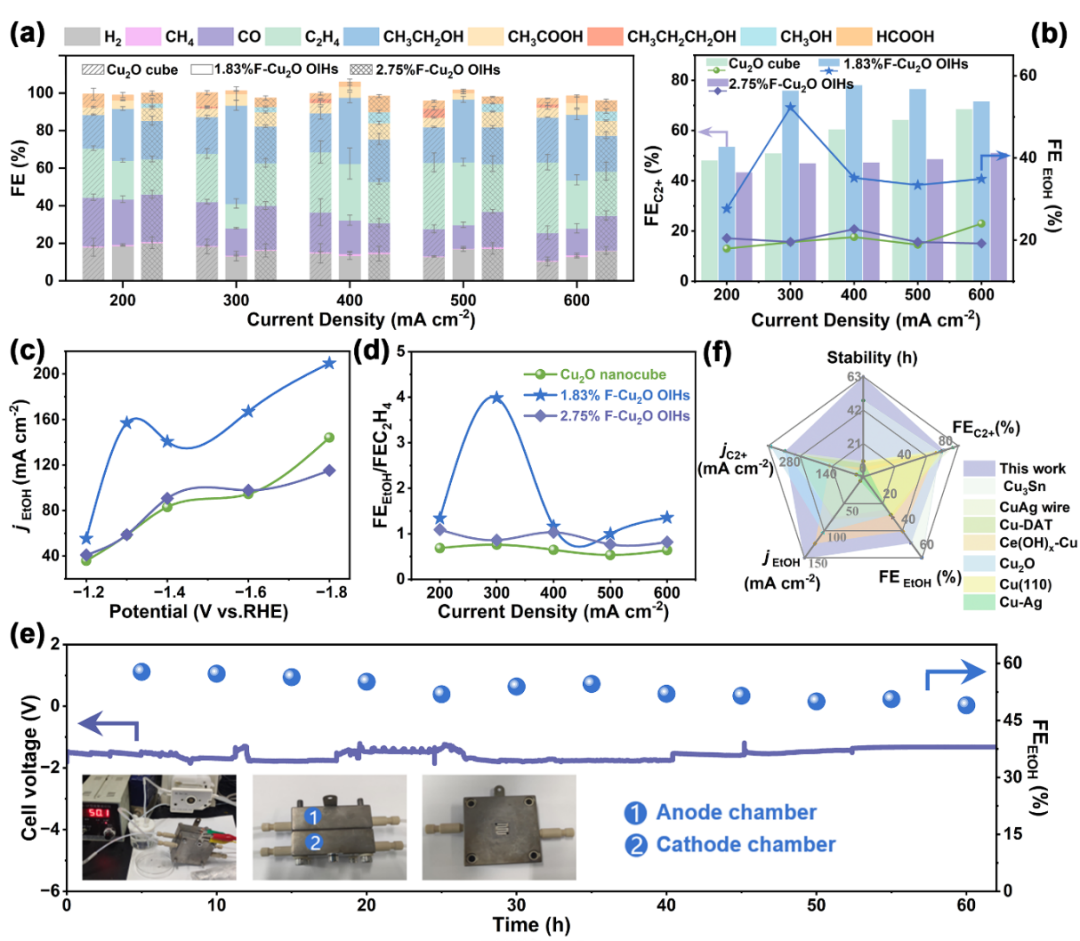
图 2:x%F-Cu
2
O
OIHs的CO
2
RR性能。(a) CO
2
RR产物分布;(b)FE
EtOH
和FE
C2+
;(c)
j
ethanol
;
(d)
FE
ethanol
/FE
ethylene
比率;(e) MEA中稳定性评估;(f)性能对比分析。
通过多方面数据展示不同催化剂在
CO
2
RR
中的性能,主要突出1.83%F-Cu
2
O OIHs在产物选择性、电流密度、稳定性等方面的优势。在产物分布方面,于100 -600 mA cm
-2
操作窗口内,C
2
H
4
和C
2
H
5
OH是主要C
2
产物,且C
1
产物形成被抑制,表明该体系对C-C偶联有利。1.83%F-Cu
2
O OIHs在
j
ethanol
及FE
ethanol
/FE
ethylene
比率上均优于对比催化剂,体现其对转化为乙醇的偏好。在长时间稳定性测试中,1.83%F-Cu
2
O OIHs在膜电极电解槽中稳定60 h且FE
ethanol
保持50%左右,催化耐久性优异。最后与现有先进Cu基催化剂比较,通过多描述符体现了1.83%F-Cu
2
O
OIHs在将CO
2
转化为乙醇方面性能卓越。
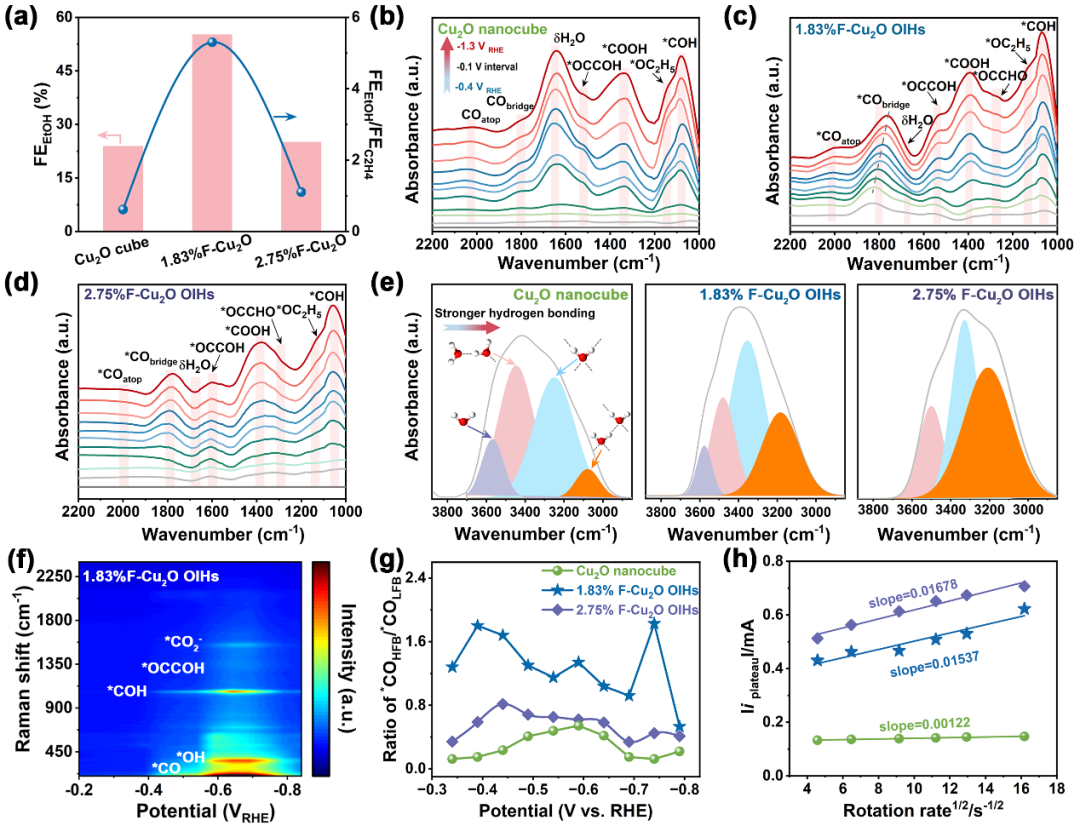
图3 加速CO
2
转化为乙醇的机理研究。Cu
2
O及x%F-Cu
2
O OIHs (a) 火山形性能趋势;(b-d) 原位红外光谱动态研究;(e)水的光谱特征;(f) 1.83%F-Cu
2
O OIHs原位拉曼光谱;(g) *CO
HFB
/*CO
LFB
比率;(h)质子扩散系数测定。
本文运用多种原位光谱技术及电化学测试,探究C
8
F
18
植入Cu
2
O实现C
2
产物逆转的机理。我们发现植入的C
8
F
18
对FE
ethanol
比率的影响呈现火山形关系。通过ATR-SEIRAS发现C
8
F
18
-Cu
2
O OIHs的*CO
bridge
信号更明显且红移,起始电位顺序体现适量C
8
F
18
对其形成的影响,同时*COH相关振动带也有规律,与*CO
bridge
相关质子化解释了乙醇形成优势。通过高斯拟合分析了约3400 cm
-1
处的宽峰以阐明水的溶剂化结构。C
8
F
18
增加使水溶剂化结构变化,氢键连接性增强。SERS检测表明1.83%F-Cu
2
O OIHs有优越的CO覆盖率和更强C-C偶联能力。RDE循环伏安曲线测试显示C
8
F
18
-Cu
2
O OIHs质子可及性更高,计算得出其质子扩散系数大幅提升,归因于更有效的H
2
O活化,这些研究从多维度阐释了C
8
F
18
植入的作用机制。
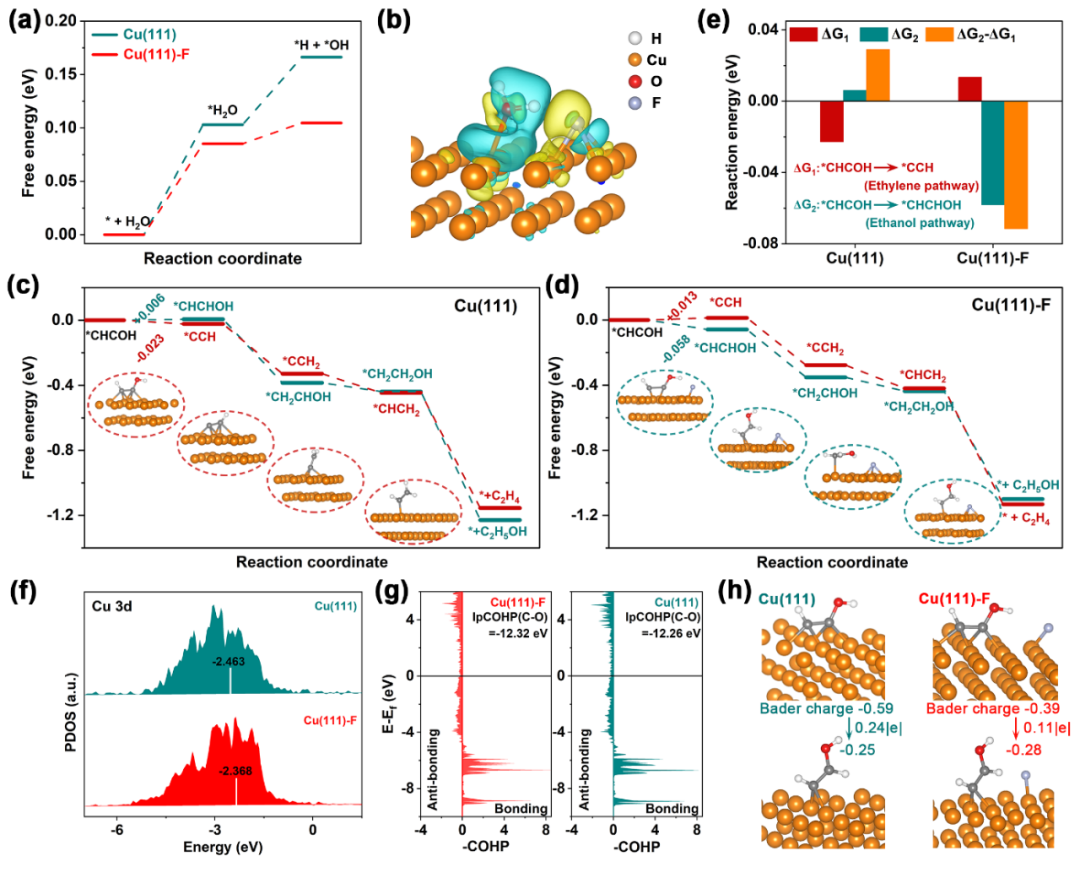
图4:DFT模拟揭示乙醇选择性增强机制。(a)水的解离能;(b)电荷差分密度;(c-d)反应路径能量变化;(e)*CHCOH加氢反应能对比;(f)轨道态密度;(g)-COHP;(h)巴德电荷转移。
本文利用DFT模拟深入探究F修饰Cu
2
O催化剂提升乙醇选择性的机制。计算显示,F使H
2
O在Cu(111)-F上的吸附能与解离能垒降低,得益于增强的氢键网络,其拉长O-H键加速解离,增加*H覆盖度以加速*CO质子化,与实验结果一致,差分电荷密度也印证此影响。模拟CO
2
在Cu(111)和Cu(111)-F上从*CHCOH分支点的反应能量变化也证实了,F植入后影响了乙醇途径的逆转。Cu(111)-F吸附*CHCOH中间体时,Cu 3d轨道在费米能级附近电子占据高,相互作用强。pCOHP分析表明Cu(111)-F上*CHCOH的C-O键更强,阻碍乙烯生成。Bader电荷分析显示Cu(111)-F的Cu-C键较弱,利于CO
2
还原为乙醇。综上,DFT模拟多维度阐释了F修饰提升乙醇选择性的机制。
本研究采用软模板法将C
8
F
18
植入Cu
2
O,制备了有机-无机杂化结构(OIHs)催化剂用于CO
2
电还原(CO
2
RR)。通过多种表征手段证实了C
8
F
18
的成功植入及对催化剂结构和性能的影响。电化学测试表明,1.83%F-Cu
2
O OIHs在CO
2
RR中展现出优异的乙醇选择性(FE
ethanol
约55.2%,乙醇/乙烯比约5.4)和稳定性。机理研究揭示,C
8
F
18
增强了界面氢键网络,促进活性氢物种生成,稳定乙醇相关中间体,推动反应向生成乙醇方向进行。DFT模拟进一步阐释了相关作用机制。
本研究充分展现了运用非共价相互作用调节二氧化碳电还原生成乙醇选择性方面的极大潜力
,为实现高效CO
2
RR制乙醇提供更有力的理论与技术支持,推动其在可再生能源转化领域的应用。
文献链接:https://onlinelibrary.wiley.com/doi/10.1002/ange.202418459
欢迎关注我们,订阅更多最新消息










