让我们重温经典,紧跟监管机构审评考虑,开发出“以患者为中心,以科学为基础”的抗肿瘤新药。
NMPA.CDE杨志敏部长分享了题为《2020年中国抗肿瘤创新药审评情况报告》,以下是全部录音、全部PPT和讲解的全部内容:



扫描以上图片二维码观看科临界所有的直播和回放。

首先,非常感谢CSCO对我们的支持和指导,特别是江泽飞秘书长在我们之前的准备中,包括我们今天的所有的题目的制定,都是我们结合在这一年的过程中,
行业发展过程中遇到的一些问题,或者我们大家在共同推进的时候,需要解决的瓶颈,来制定的既有创新前瞻性的考虑,又有务实解决当前问题的思考
,所以形成了今天下午的专题,这里我就不再对每一个具体的专题来介绍了,后续我的同事也会逐个向我们在座的、线上线下的各位同道来分享,这里我按照CSCO给我们也是布置的一年一度的一个要求,我代表整个CDE的抗肿瘤团队向各位专家、给位同道来汇报一下
2020年我们中国抗肿瘤创新药审评情况报告。

我的报告还是从
审评工作的情况,审评的考虑以及挑战
这几个方面来和大家分享:

首先第一个,我们还是来介绍一下
2020年批准上市的抗肿瘤这些新药的情况
,今年批准的抗肿瘤新药有几项特点:第一,咱们可以看到最右边的这些从数量上来看
19个
,其实数量并不重要,重要的是它代表了什么,我们可以看看右边的这个临床意义,我们实际上在今年批的里边,
第一个有咱们国内第一个ADC药物批准了,第二个在小细胞肺癌里边,长久以来难以突破的领域,获得了免疫性治疗的批准,第三个有我们国产的、自主知识产权的、第三代TKI的新药获得了批准,另外,更加重要的是除了我们非小大家都公认的非小细胞肺癌、乳腺癌等等这些大瘤种以外,我们有了肝癌、食管癌、胃癌这些是我们自己高发的这些肿瘤的免疫治疗药品的批准,同时,我们也有一些,比如说,像曲妥珠单抗这样的生物类似药在中国和欧盟都同步获得了申报和批准,这些都是我们在批准里边获得的应该说是对我们肿瘤患者带来的新的治疗手段,甚至有的产品改变了治疗领域的实践
。
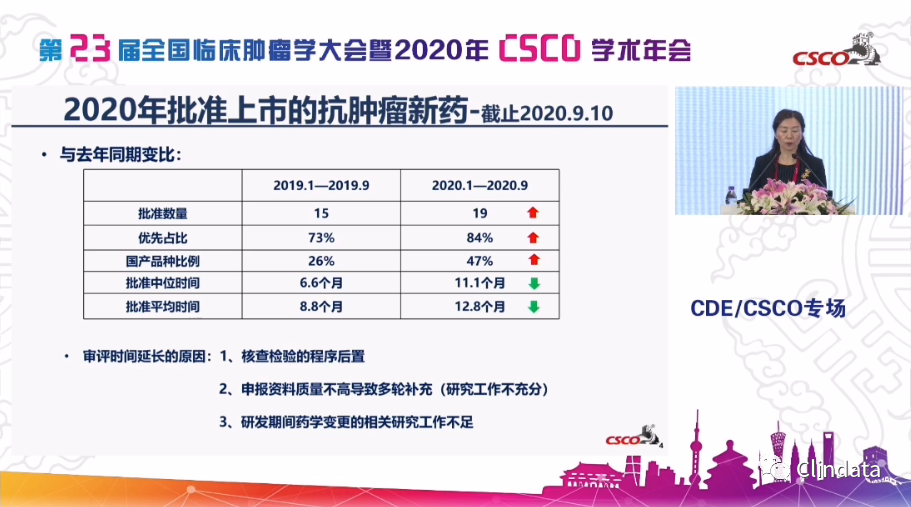
让我们从抗肿瘤新药进一步看到,今年总体的、在疫情情况下,批准的数量同期相比较,因为我们每年都是九月份,对同期比是升高的,
从15个到19个
,同时,优先的品种由原来的
73%上升到现在的84%,还有一个我们也是觉得可喜的这么多年积累的,就是国产的获批的比例已经将近50%,但是,另外一个让我们值得思考的就是中位的批准时间和平均时间较去年是有所下降的,当然为什么会这样的情况,我们也进行了逐个的分析:一方面,是程序的问题,大家也知道我们新法规的实施,这个问题未来会得到妥善的解决;第二个和第三个实际上对我们整个药物研发来说,也是大家关注的,在创新药的研发过程中,大家常常给予我们的作业,从写作业的方式去做,而不是从一个产品整体的规划来考虑,所以等到NDA批准上市的时候,出现一些资料的问题,研究不充分等等需要多轮的发补,再去讨论,耽误了一些时间,所以到了我们时间还是有些问题。
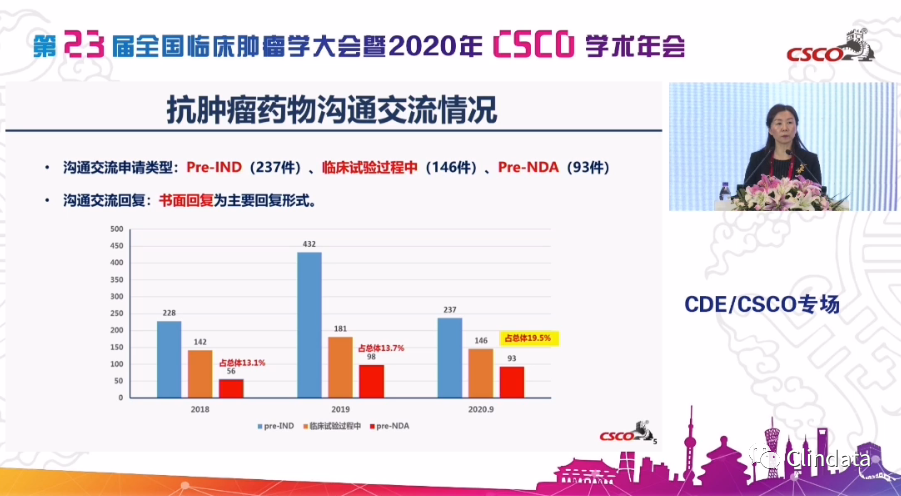
另外,
在三个方面沟通交流,因为这是创新药非常重要的部分,我们也可以看到,实际上整体来看,这几年沟通交流的数量基本上是持平的,特别是2019年的时候,因为有个默认许可实施,所以当时Pre-IND沟通交流比较多,今年大家这方面比较成熟,略有下降,但是NDA的沟通交流明显增加,所以这点意味着我们前几年我们整个行业和专家共同努力的创新药会在未来的,在今年未来的几年不断的开花结果,所以这就是我们在沟通交流完成的工作。
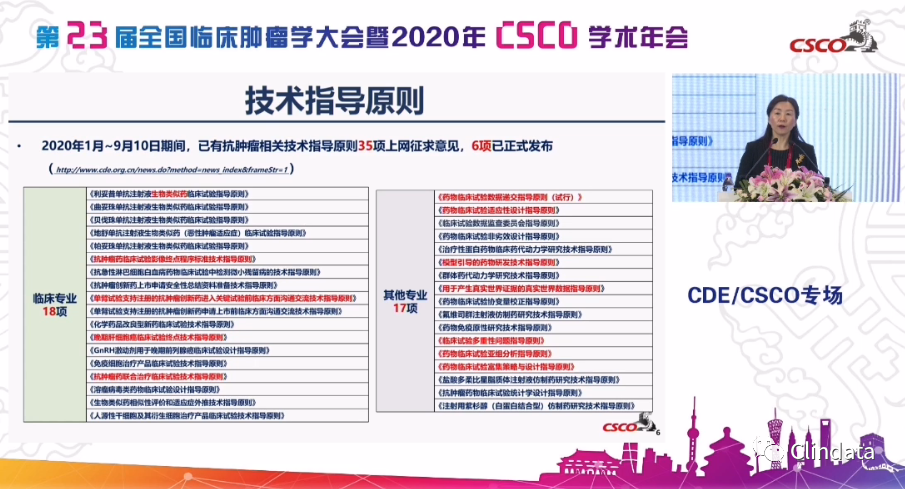
另外一个非常重要的就是技术指导原则体系的建立,因为这也是我们GRP,良好的审评管理体系里边非常重要的质量标准的体系,今年整个药审中心应该说是指导原则的大年,光和我们肿瘤相关的指导原则就有
35项
,而其中和临床相关的就是
18项
,还有我们统计学等等、临床药理相关的有
17项
,这些指导原则涵盖了在我们新药研发过程中大家特别关注的这些问题,有逐个的,比如说我们肝癌指标,有共同的比如说单臂研究设计的沟通交流,还有一些既有生物类似药个例的指导原则,也有一些非常前瞻性的比如说模型引导的药物研发,实际上这些指导原则的建立将推动我们整个的在抗肿瘤领域在创新药研发的一个标准体系的建设,在这里我也特别感谢我们线上和线下的各位专家、同道在我们指导原则起草、制定和征求意见过程中给了我们非常多的建议,非常多的反馈的一些很好的思路,所以这也对我们未来的指导原则的制定会奠定一个很好的基础。

第二个也是我们大家非常关注的,当我们在看我们取得的一些成果,或者说我们在获得越来越多产品批准的时候,实际上在我们这一年的工作过程中,我们还发现了一些焦点的问题,这些问题也是我们常常需要思考的,那这些问题集中在那些方面呢?

第一个非常主要的方面就是制定一个临床研发的整体规划,从前面的数据我们可以看得出来,虽然我们国内就像昨天我们在CSCO的大会上,我们的专家也说到,我们从跟跑慢慢的越来越多的进入领跑,但是我们实际上从照猫画虎的状态,慢慢要前进到要理解很多创新设计它下面的科学道理,这样的话我们才能基于科学去做我们的研究设计,所以在开始临床试验初始的时候,特别是第一个人体剂量的设计的时候,我们一定要根据产品的作用机制、非临床前的产品预测和有可参考的数据来建立我们研究设计的模式,
至于我们现在是希望用规则指导的原则,比如说3+3,快速滴定,还是我们用模型,或者说我们加入更多统计学原则思考的方式,这个都是我们需要讨论的
,另外,在早期的研究过程中,我们更多希望加入一些生物标志物,把更多更灵活的科学的元素融到早期研究中,为我们后面的打好基础,然后第二个方面就是剂量探索,实际上拓展研究是我们进入关键研究非常重要的方面,所以在这里我们更多的也是根据它的作用机制,而且已经获得初步疗效数据和临床急需的程度,包括它疗效有多好,来讨论它的研究设计和病例数,而不是我们简单的Simon two stage,Stage I做什么,Stage II做什么,究竟是做30例,40例还是50例,60例,大家不要拘泥于这些数据表面,而要根据它的实际情况,或者基于它的已经获得的数据,大家来探讨思路,就是研究设计。

另外一个常常被忽略的就是特别我们国内的企业有时候,一心一意的就做关键研究,常常会把一些关键的药理学数据忽略了,比如说药物相互作用的问题,物料平衡的问题,特殊人群的问题,这也是常常制约我们在新药研发过程中使患者能够得到获益和风险权衡的一个数据,另外,特别在抗肿瘤药物研发过程中,我们也希望在早期的研发过程中,特别是进入了一些我们已经获得了很好的数据过程中,我们引入青少年的一些患者进入,这样推动我们儿童用药,另外,一个安全性数据的暴露以及风险控制计划,这些都是我们在整体的规划过程中特别关键和重要的。

第二个,就是我们进入关键研究的时候,企业或者本身的特点来考虑,我们在哪个适应症上突破,大家不要扎堆儿,一看国外做肺癌,我们就做肺癌,国外做肠癌,我们就做肠癌,而是要根据我们产品本身的特点、医疗实践来确认,是做罕见瘤种入手,还是高发瘤种入手,我们从末线,还是从前一线,包括用单药,还是用联合,当然用联合我们都是根据药物的作用机制来选择什么样的联合,还是我们直接和单药对照,这些都是要根据基本的情况来确定。
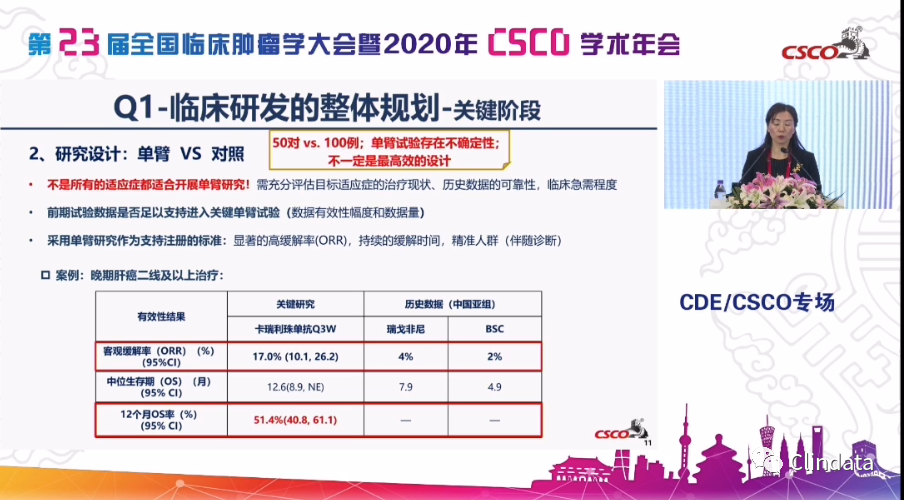
另外,特别重要的一条是用单臂还是对照,我们最近也可以看到,我们出了两个与单臂相关的指导原则,是不是大家都认为就做单臂就能获得批准,是不是最高效的,其实未必。50对的对照和100例的单臂,实际上它们对有效性,安全性的一个判断的权重是不一样的,因此并不是所有的适应症都适合用单臂研究,我们希望在高的一些缓解率,缓解持续时间长,特别是精准的人群里开展项目研究,我们从下面可以看到,我们在免疫治疗里,我们的缓解率不仅高,在高的缓解率的同时,我们还想看到好的持续缓解时间,这些都是我们能够获得批准的考虑。
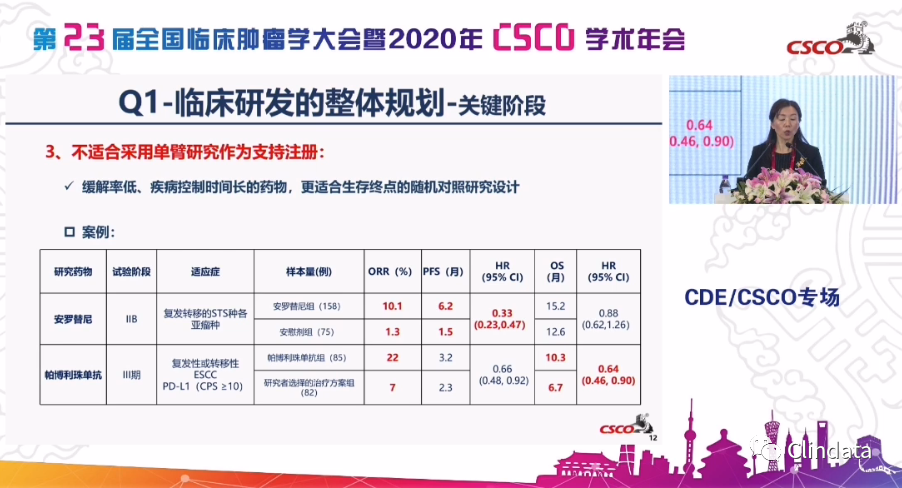
同时,对一些缓解率低的、持续时间又长,甚至有生存获益的,比如说我们举了两个例子,像抗血管,或者说我们的免疫治疗,可能ORR不是它最主要的获益的时候,我们继续要考虑PFS,我们要考虑OS等等,用更多的研究设计去看到它的优势或好处,而大家不是一想到批准上市就用单臂,这是我们在今年的过程中大家特别关注的。
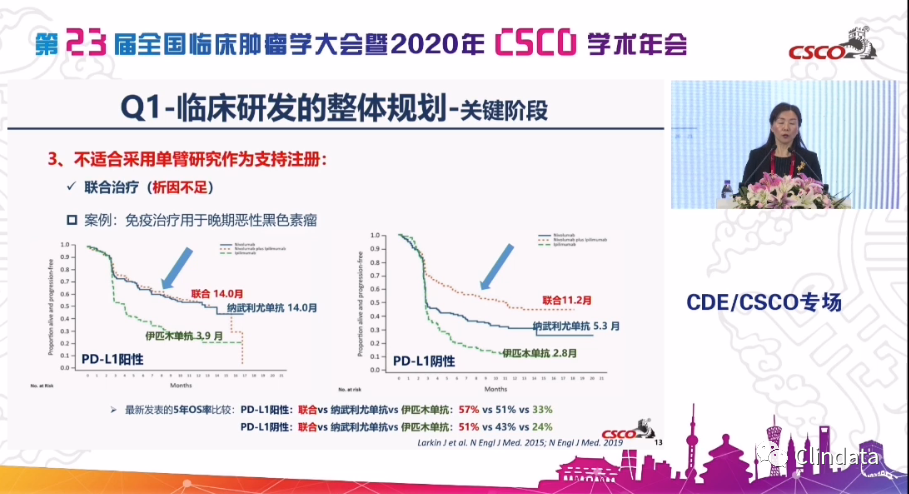
另外还有一个特别关注的就是联合治疗。基本的所有的大家都希望在单臂的同时,也看到了国外也批准了几个,特别是我们的双免疫,大家也知道我们在双免疫,在肺癌、黑色素瘤,最近刚出的在胸膜的间皮癌都获得了批准。所以希望用单臂,实际上我们还是希望提醒大家,我们可以从这张图里看得出来,实际上当我们的人群选择不好的时候,我们是分不出单药的贡献的,当然我们可以看比如恶性黑色素瘤就能看得出来,我们只做一个联合的话,看两个联合很好,但我们实际上看左边的图,当我们的PD-L1假如说高表达的情况下,我们单药和联合用药实际上差不多的,虽然我们长期随访5年的数据可以看得出来,它们会分开一些,但是这个析因在单药治疗和联合治疗就变得非常重要。所以,反过来说对于联合治疗,如果没有充分析因的情况下,用单臂也不是特别合适的一种选择的模式。

同时更得大家关注的是一到关键研究,目前很多人认为我们能不能做个ORR的两组比较,但是ORR毕竟是个点估计,而是时间点的获得的缓解数据,在如果非常确定的ORR、PFS和OS的相关性,这是第一个前提,第二个获得了非常好的ORR的一个情况下,通常我们不主张,你看我们最近三线GIST的数据,大家就看得出来,阿泊替尼实际上ORR翻了倍了,PFS没有升高,实际上我们在结直肠癌也看到这样的情况,包括原来的肝癌布立布尼也是,ORR由原来的5%升高到25%,结果最后PFS和OS都没有提高,所以我们如果没有充分的证据的情况下,我们把ORR作为单终点不合适,但是我们有数据的存在建立了相关性的时候,ORR\PFS\OS是可以作为一个可以印证的双终点来同时作为我们观察的,特别是我们下面举得这个例子,如果这两个药我们都把PFS和OS作为一个共同终点,在我们都没有获得OS的情况下,如果PFS仅仅延长了1个月,HR是0.7,可能这个时候我们要等OS了,但是对于一个临床急需的,比如说现有的疗效不是很好的情况下,我们PFS已经延长了5个月,HR0.3,在这种情况下也许就可以采取一种更创新的监管模式,我们就可以提前来申报,再等OS,所以,我们在讨论一个问题的时候,原则是基于它的数据来考虑的,所以我们沟通交流的时候,我们常常有企业来问说,哎呀我们可不可以用这样的数据来获得批准,其实我们讨论的首先是规则,但是规则下面还是有数据,比如说刚才也说到,如果我们现有的ORR只有10%或者20%,这个时候我们的ORR已经变成了70%,本身这么巨大的ORR的提高,或者伴随着持续时间长的,我们没有PFS和OS的数据,我们都认为是很好,但是如果仅由5%-10%的ORR基础数据提高到20%-30%,可能这时候我们要再问一个问题,这样的ORR能不能转化为PFS,所以,我们还是基于数据基于科学的注册策略,而不是我们一问就问ORR是不是可以作为标准,这就是我们在创新过程中要讨论的。




