
叔丁基亚磺酰胺在手性胺类药物合成中的应用
Applications of tert-Butanesulfinamide in the Synthesis of Chiral Amine Drugs
朱怡君,宦玉亮,冷卫东,伍成祥,王 兵
(常州制药厂有限公司,江苏常州 213011)
手性胺类药物都含有手性胺官能团,其合成方法主要分为传统的外消旋体拆分和不对称合成2种。
不对称合成主要包括2类方法:
①以Mitsunobu反应为代表的SN2反应。
该法在药物合成中应用广泛,但该类反应会产生大量的三苯氧膦,且有时构型反转不完全导致产物的光学纯度不高;
②亚胺的不对称还原。
依据氢的来源不同又可分为2种方法:一种是在金属催化剂和手性配体催化下加氢还原,但手性配体及催化剂均较昂贵,限制了其在工业化生产中的大规模应用;另一种是手性辅剂诱导,即醛(酮)先与适当的手性辅剂缩合形成亚胺,再经还原,最后脱除手性辅剂的辅助部分。辅剂诱导合成手性胺类药物,应用最广的是甲基苄胺。
1997年,Ellman课题组通过固相合成首次得到了物理、化学和光学性质均稳定的光学纯叔丁基亚磺酰胺,其作为合成手性胺的新型辅剂引起了研究人员的广泛关注(图1)。

叔丁基亚磺酰胺作为手性辅剂,主要有以下优点:
(1)易于与各类醛或酮形成亚胺;
(2)由于叔丁基亚磺酰基的活化作用,生成的亚胺亲电性更强,更易于与亲核试剂反应或被金属硼试剂还原,诱导出高的非对映选择性;
(3)在加成产物中,叔丁基亚磺酰基也是一个很好的保护基团,能够耐受反应中的强碱、过渡金属等;
(4)酸性条件下,叔丁基亚磺酰基容易脱去,所得盐酸盐可用醚类溶剂纯化,收率几乎定量(图2)。

本文主要综述近几年来叔丁基亚磺酰胺在手性胺类药物制备中的应用情况。
1、卡巴拉汀的合成
卡巴拉汀(rivastigmine,1),化学名为(S)-N-甲基-N-乙基-3-[(1-二甲胺基)乙基]氨基甲酸苯酯,是诺华公司研发的一种乙酰胆碱酯酶抑制剂,临床用于治疗老年痴呆症,1998年经EMA批准上市。目前已有多篇文献报道了1的合成路线,这些方法大多先合成外消旋体,再经手性拆分得1。陈卫民等以间甲氧基苯甲醛与(R)-叔丁基亚磺酰胺缩合生成亚胺2,然后与甲基碘化镁加成得化
合物3,再依次经脱保护、Eschweiler-Clark甲基化及酚羟基脱保护得关键中间体4,最后与甲乙胺基甲酰氯酯化得1(图3)。该法合成手性胺产率高,ee值大于98%。2010年,该课题组还采用上述方法合成了1的12个类似物。

2011年,Arava等以间羟基苯乙酮与甲乙胺基甲酰氯酯化,再与(S)-叔丁基亚磺酰胺缩合、还原、水解,得到关键手性胺5,最后再甲基化即得1(图4)。该法总收率达76%,且ee值100%。

2、硼替佐米的合成
硼替佐米(bortezomib,6),是美国Millennium公司研发的蛋白酶体抑制剂,于2003年获美国FDA批准上市,临床用于治疗多发性骨髓瘤和复发性难治性套细胞淋巴瘤。其合成难点在于α-氨基硼酸酯中间体的合成。目前专利路线以2-甲基丙烷硼酸为原料,以(1S,2S,3R,5S)-(+)-蒎烷二醇为手性配体缩合成硼酸酯,再在无水氯化锌催化下插入氯亚甲基,接着进行胺的亲核取代、脱三甲基硅基和氨基酸偶联,最后和哌嗪酸偶联得6。该法原料(1S,2S,3R,5S)-(+)-蒎烷二醇价昂,且氯甲基化操作条件苛刻,收率较低,不易工业化生产。2008年,Ellman课题组报道了一条新路线(图5),从(R)-叔丁基亚磺酰胺出发,通过双联频哪醇酯(B2Pin2)的不对称加成得关键中间体7,大于98∶2。该路线成本低廉、便于生产。2012年,中国人福医药申请了类似方法的专利。

3、盐酸西那卡塞的合成
盐酸西那卡塞(cinacalcet,8),化学名为N-[(1R)-1-(1-萘基)乙基]-3-[3-(三氟甲基)苯基]-1-丙胺盐酸盐,2004年经美国FDA批准上市,临床用于进行透析的慢性肾病患者的继发性甲状旁腺功能亢进症及甲状旁腺癌患者的高钙血症治疗。其一般制备方法为(R)-1-萘乙胺与3-(3-三氟甲基苯基)丙醛还原氨化,有专利用萘乙酮与(R) -叔丁基亚磺酰胺缩合、还原,再与3-卤丙炔反应得化合物9,再与间卤三氟甲苯偶联得化合物10,再经还原、成盐即得8(图6)。

2012年,Arava等同样以萘乙酮为起始原料,与(R)-叔丁基亚磺酰胺缩合、还原,再与1-(3-卤素丙基)间三氟甲基苯亲核取代得化合物11,最后水解、成盐即得8(图7)。

4、琥珀酸索利那新的合成
琥珀酸索利那新(solifenacin succinate,12),是安斯泰来公司研发的一种M受体拮抗剂,2004年被EMA和美国FDA同时批准上市,临床用于膀胱过度活动症患者伴有的尿失禁和/或尿频、尿急症状的治疗。其合成方法大多都以(S)-1-苯基-1,2,3,4-四氢异喹啉和3-奎宁环醇进行拼接,所用异喹啉中间体须先制备消旋体再经拆分获得。2014 年,Babu等用2-(2-溴乙基)苯甲醛和(R)-叔丁基亚磺酰胺经缩合、与格氏试剂加成、亲核关环,再水解制得关键中间体(S)-1-苯基-1,2,3,4-四氢异喹啉(13),再和3-奎宁环醇缩合、成盐得12(图8)。

5、甲磺酸雷沙吉兰的合成
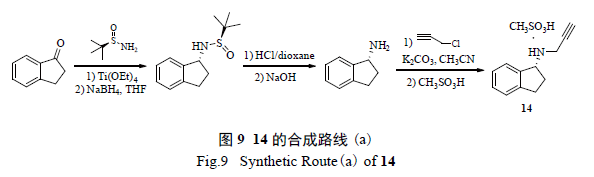
甲磺酸雷沙吉兰(rasagiline mesylate,14)是以色列梯瓦公司和丹麦Lundback公司联合研发的第2代选择性单胺氧化酶-B(MAO-B)抑制剂,2005年在欧洲上市,临床用于治疗帕金森病。其合成方法很多,传统方法是采用酒石酸拆分。郭舜民等用(R)-叔丁基亚磺酰胺与1-茚酮生成亚胺,不经分离直接还原,再水解得手性胺,再经取代、成盐即得14(图9)。该法收率较高,终产物ee值96%。
2013年,能特科技股份公司也报道一条类似的路线,也是用(R)-叔丁基亚磺酰胺为手性助剂,区别在于还原所得亚胺先与3-卤丙炔缩合,再水解、成盐即得14(图10)。该法所得14光学纯度高达99.5%。






