
前文依托考昔为例:适度区分力的溶出方法开发与验证指出,任何具有区分力的溶出方法的开发需要以下步骤:
接下来的这篇文章是对该思路的完美诠释!该文章由达沙替尼片原研公司——百时美施贵宝发表。该溶出方法开发于达沙替尼片一期临床结束后、二期临床开始前,约在2003年。该文描述了在新药研发中如何系统的开发溶出方法。
难溶性药物是在研小分子药物的主流。其仿制药处方工艺开发的难点在于找到“目标曲线”。谢沐风老师为仿制难溶性药物设计的溶出路线图为:原研如没有一条曲线能在45~120min达到85%,则可适当放宽试验条件(以原研制剂样品批内/批间精密度为准,加大转速或加入低浓度表面活性剂)、且无突释和拐点,这样的曲线通常被认为是最具体内外相关性,也是最理想的曲线。这是剖析原研,找到原研的那一根“筋”,指导仿制药处方工艺的开发。
达沙替尼(Dasatanib, BMS-354825)是一种强效的酪氨酸激酶多靶点抑制剂。为继尼洛替尼之后另一个用于伊马替尼耐药和不耐受的慢性粒细胞白血病(CML)慢性期的药物。与针对Bcr-Abl融合蛋白单靶点的伊马替尼和尼洛替尼相比,达沙替尼属于多靶点药物,对5种关键性致癌酪氨酸蛋白激酶,即BCR-ABL、SRC、c-KIT、PDGFR和Ephrin(EPH)均有作用。
该产品最早由百时美施贵宝公司研发,2006年6月在美国获准上市,11月在欧盟上市。我国已进口,商品名施达赛®。国产药品为正大天晴,于2013年上市。达沙替尼为BCSⅡ类。文献报道其溶解度为0.0128mg/ml,logP为1.8,pKa值(强酸)为8.49,pKa值(强碱)为7.22。
运用实验设计原则开发溶出方法:达沙替尼片
为了支持二期临床,溶出方法必须快速完成开发。最终确定的溶出方法为:1000mL,pH4.0醋酸盐缓冲液 1%Triton X-100,USP桨法,60 rpm。
DOE通常用来设计开发制药生产工艺。从分析方法方面来看,DOE可以用于方法开发和验证,建立控制空间,目的是更好的理解关键参数及其细小变化带来的影响。现在DOE已经应用于HPLC、GC等方法开发。但是尚未用于溶出方法的开发。
为了加快上市,同时最终上市的规格未确定,我们选择了常规的制粒工艺。达沙替尼为BCSⅡ类(低溶高渗),溶出度是该处方的一个关键质量属性。开发一个具有区分力的溶出曲线,评价批间变异,比较不同规格,以支持生物豁免。
为了加快进度,溶出方法开发需要:
可利用的资源有限,因为原料药有限;
时间不超过3个月,包括4周稳定性数据。
系统的方法开发可以发现关键参数及其相互关系。实验设计为:在一个6杯溶出仪中3个不同条件,每个条件2片。这个设计用于摸索溶出方法的设计空间。当确定最终的溶出条件时,片子的数量增加,以保证把片与片之间的变异考虑在内。这种方法的优点在于:
开发目标为溶出慢(20min时,溶出不超过85%)并且全面(60min时,超过95%)。如前所述,最终上市规格未确定,溶出方法应覆盖20-150mg。本文采用150mg规格开发溶出方法。如果用低规格的片,可能最高规格(150mg)在选定溶出介质中并不能完全溶出。
实验计划是根据溶解度曲线制定的。API溶解度随pH升高显著降低。当pH2.6,溶解度为18.4mg/mL;pH4.5,溶解度为0.08mg/mL;pH7.0,溶解度低于1mg/mL。同时它的碱性pKa值为6.8和3.1,和弱酸性pKa~10.9。这表明,溶出介质pH的微小变化将导致溶出曲线的显著差异,尽管满足漏槽条件。药典中的溶出条件(pH1.2,4.5,6.8)不合适。pH1.2介质中,10min溶出超过90%,溶出特别快。pH4.5介质中溶出不完全,60min溶出小于40%;pH6.8介质中,60min溶出小于2%。pH对溶出曲线具有关键影响,基于此,设计以下实验。实验计划见图1。
为了进行二期临床实验和商业化生产,开发了普通的制粒工艺,制备了薄膜包衣片,包括20、50、70、150mg规格。最初产品开发时微晶纤维素全部内加。在开发过程中部分外加以提高颗粒的可压性。这两种处方量完全相同,仅在MCC的内、外加上不同。溶出方法学开发使用了这两种处方。
pH3.0-4.0柠檬酸盐储备液 配制柠檬酸盐储备液(pH3.4):9.8g一水柠檬酸和1.0g二水柠檬酸钠溶于1L去离子纯化水中,用氢氧化钠或磷酸调节pH至3.4(±0.05)。其他pH值的柠檬酸盐缓冲液,使用磷酸或氢氧化钠将pH值调整至±0.05以内。
pH4.0-4.8醋酸盐储备液 配制柠檬酸盐储备液(pH4.5):3g三水醋酸钠和14mL 2N醋酸溶液,溶于1L去离子纯化水中,用冰醋酸调节pH至4.5(±0.05)。其他pH值的醋酸盐缓冲液,使用冰醋酸或氢氧化钠将pH值调整至±0.05以内。
加入表面活性剂的介质配制 按体积比加入到目标pH介质中,充分溶解。如果需要,检查和重新调节pH。
如无说明,采用USP二法、60 rpm、1000mL、37℃。溶出曲线取样点10、15、20、30、45、60min。溶出无穷大点为200rpm 搅拌1h。如无说明,每个溶出条件用2片(每次3个条件,6个溶出杯)。
溶出介质的pH在溶出方法中是非常关键的,有两个原因。第一,可以发现pH作为溶出主要驱动力的范围。第二,溶解度曲线可以发现潜在的目标溶出条件,它是在溶出平衡的条件下测的,得到的溶解度并不能反应溶出介质的表观溶解度。
6个溶出杯,3个pH介质,1片/杯(150mg 原处方薄膜衣片),2个溶出杯/pH介质。如图2,这种方法可以快速开发溶出方法从pH3.0至4.4。图2可以看出,当溶出介质pH升高时,溶解曲线越来越慢。数据见表1。可以分为3个不同的区域。第一个区域(pH小于3.4),溶出迅速、完全。第二个区域(pH3.4-3.6),可以接受的曲线,20min溶出少于80%;60min溶出大于95%。这些结果与pH溶解度一致,这表明pH3.4 与150mg规格的漏槽条件非常接近。第三个区域(pH3.8至4.4),溶出不完全。溶出终点小于95%,视为未达到漏槽条件。
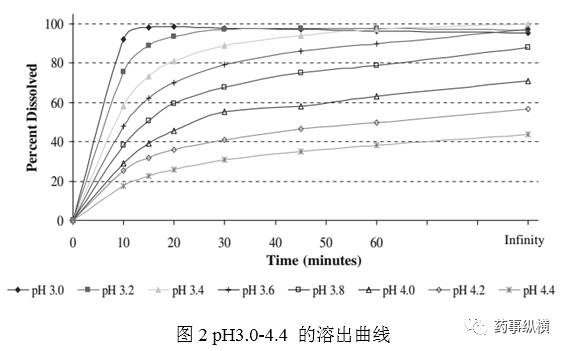
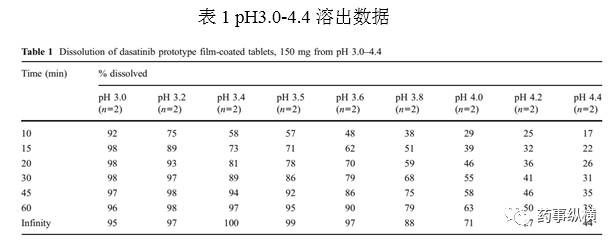
实验B,第一区域pH3.0-3.4。转速降为50rpm,以降低溶出,使20min溶出不超过80%。结果如图3所示,表明比较慢的溶出曲线在溶出杯中会有明显的堆积。溶出无穷大终点比60min提高了10-20%。
实验D,第二个区域pH3.4-3.6,介质的pH就能够直接控制溶出。表1的数据表明当pH增加0.2时,每个点的溶出约降低10%。pH的微小改变导致溶出较大的改变,表明依赖于的溶出方法耐用性不佳,也不适合用于质量控制。
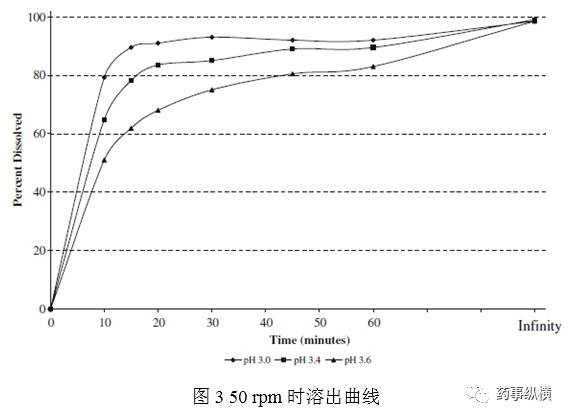
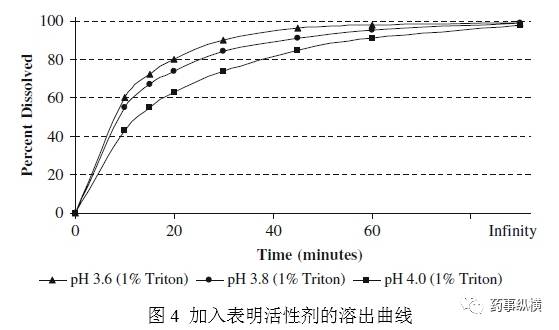
实验E评价了pH介质和表面活性剂在第三个区域的共同作用。基于pH研究数据,选择pH3.6-4.0,加入最少的表面活性剂,达到充分溶解,这样溶出介质形成的泡沫最少。在耐用性研究时,片剂在pH4.8醋酸盐SLS介质中溶出不完全且变化大。有些批次在60min时溶出少于85%。在不同pH介质中加入SLS,会导致达沙替尼相变,最终形成一种粘性的非晶相。这说明SLS不合适。筛选其他几个非离子型表面活性剂包括泊洛沙姆、溴化十六烷基三甲基铵、Triton X-100。最终选择TritonX-100,因为其使用方便,易于获得。图4表明1%的Triton X-100 pH3.8介质得到了目标曲线。同时pH3.6、4.0也同样获得了目标曲线。这个方法在pH变化时耐用性良好。
至此,合适的条件已经开发出来。为了测试其区分力,首先要开发一个新处方。
因为原料药生产批次少,对其粒度分布了解太少,处方修改以满足使用更大的原料药粒径。15%MCC外加,代替全部内加。只改变了处方的添加方式。处方改变需要开发更多的方法,但是,只有60片可用于实验。为了加快审评,需要减少方法开发的时间(包括验证)。为了克服这些挑战,在利用原处方片得到的溶出数据的基础上设计了一系列实验。
表1中的溶出终点,代表了达沙替尼固有的溶解度,不管处方如何。如此,在低于pH3.4的溶出介质中,溶出快速、完全。但在高于pH3.8的介质中,溶出仍然不完全。于是,新处方的溶出曲线在pH3.4-3.6来评估处方改变的影响。在图5中可以看出,pH3.4、3.5的溶出快于目标(20min溶出超过85%)。pH3.6能够满足目标溶出曲线。但是在表1中,pH3.8溶出终点明显下降,只有88%。从耐用性方面看,pH对溶出的影响增大,方法耐用性不佳。下一步同时优化pH和表面活性剂。
在优化表面活性剂之前,采用pH3.2,3.4,3.6介质,降低转速至50rpm。结果让我更加确认商业化处方不适合50rpm。前面原处方采用50rpm时,溶出曲线变慢,但是溶出杯底部有堆积。假设新处方不会有堆积。假如加入沉降篮或者采用USP 1法(实验C)也可以防止堆积。采用pH3.2,3.4,3.6介质,加入1%Triton X-100,验证这两个假设。结果表明堆积无法避免,在转速增至200rpm时,溶出有较大增加(从10%-40%)。
原处方在pH3.8介质1%Triton X-100得到目标溶出曲线。如其pH研究中,稍微缓慢的溶出曲线,适合商业化处方的pH为4.0-4.8,加入1%Triton X-100。图6表明pH4.0、4.2均是可接受的溶出曲线。这一pH范围内耐用性良好。
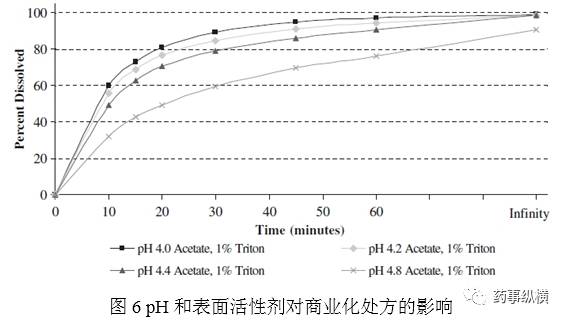
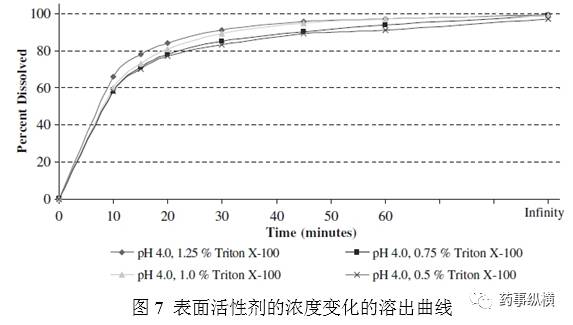
优化表面活性剂的浓度,体积比分别为0.5%、0.75、1.25%。图7表明使用0.75、1.25%,与1%的溶出曲线的相比,计算f2因子,分别为72、68,表明在±0.25%范围内,耐用性良好。表面活性剂±2.5mL/L是可以接受的。
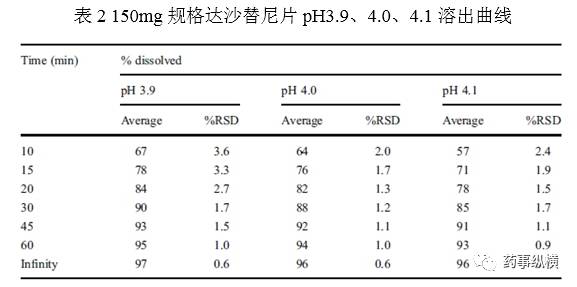
由三位实验人员进行了中间精密度实验。每人都有开展溶出实验的资格证,采用1%Triton X-100 pH3.9、4.0、4.1介质,每个介质2片。每人独立配制介质,使用不同的溶出仪。每个pH介质综合分析,n=6。结果表明方法耐用性良好。pH3.9介质的f2值为80,pH4.1介质的为65。这一结果进一步确认了方法开发时的趋势。表2显示,RSD非常低,精密度良好。整个系统开发过程仅仅使用了50片。
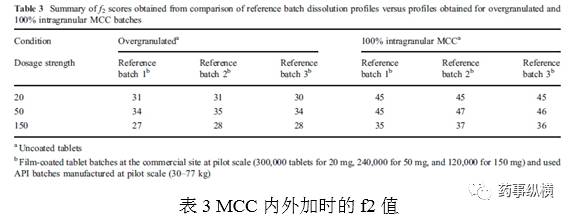
制粒过度和MCC全部内加都影响到颗粒的可压性,对片剂的质量有直接的影响,即降低溶出。溶出方法的区分力就采用这两个条件来进行验证。过度制粒是制粒时加入过量的水,混合时间延长,影响了颗粒的可压性。参照批次为按照商业化规模正常生产的3批,20、50、150mg各一批。一批商业批和一批过度制粒批一起开展溶出实验。图8为150mg规格的溶出曲线。表3为f2值。溶出方法具有区分力。
外加15%MCC可以提高颗粒可压性。湿法制粒时,内加MCC会降低可压性,外加部分MCC可以弥补。结果同样在图8、表3。证明溶出方法具有区分力。
本文开发了达沙替尼片耐用的、具有区分力的溶出方法。根据指导原则,系统的评价了溶出介质的pH、表面活性剂类型和浓度。根据实验设计原则,系统地筛选方法这些实验,利用有限的时间和资源,完成了项目的关键里程碑。
受介质pH值决定的溶出方法耐用性不足,不能够用于日常监控。在溶解度不满足漏槽条件的pH值介质中,需加入表面活性剂。研究结果也表明pH4.0介质,溶出足够缓慢、完全、可重现,耐用性良好。
50rpm桨法和100rpm篮法均能造成底部堆积。溶出方法的区分力也进行了验证。
1.Goupy J. What kind of experimental design for finding and checking robustnessof analytical methods. Anal Chim Acta. 2005;544:184-90.
2.Dourdat-Deschamps M, Daudin J-J, Barriuso E. An experimental design approach tooptimise the determination of polycyclic aromatic hydrocarbons from rainfallwater using stir bar sorptive extraction and high performance liquidchromatographyfluorescence detection. J Chromatogr A. 2007; 1167:143-53.
3.Sivakumar T, Manavalan R, Muralidharan C, Valliappan K. Multicriteria decisionmaking approach and experimental design as chemometric tools to optimize HPLCseparation of domperidone and pantoprazole. J Pharmaceutical and BiomedicalAnalysis. 2007; 43: 1842-8.
4.Destandau E, Vial J, Jardy A, Hennion M-C, Bonnet D, Lancelin P. Robustnessstudy of a reversed-phase liquid chromatograpic method for the analysis ofcarboxylic acids in industrial reaction mixtures. Anal Chim Acta. 2006;572:102-12.
5.Araujo P, Couillard F, Leirnes E, Ask K, Bøkevoll A, Frøyland L. Experimentaldesign considerations in quantification experiments by using the internalstandard technique: cholesterol determination by gas chromatography as a casestudy. J Chromatogr A. 2006; 1121: 99-105.
6.Donato P, Stancanelli R, Calabrò ML, Tommasini S, Cutroneo P, Guardo M, et al. Optimization of a LCmethod for the enantiospeparation of a non-competitive glutamate receptorantagonist by experimental design methodology. J Pharmaceutical and BiomedicalAnalysis. 2006;42:543-8.
7.International Conference on Harmonisation of Technical Requirements forRegistration of Pharmaceuticals for Human Use. Pharmaceutical Development Q8(R1); Current Step 4 version dated 13 November 2008.
8.Bioavailability and Bioequivalence Studies for Orally Administered DrugProducts-General Considerations; Guidance for Industry; US Department of Healthand Human Services, Food and Drug Administration, Center for Drug Evaluationand Research,Washington, D.C., March 2003.
本文转自药事纵横,转载仅为交流学习,如有版权问题请联系小编删除。











