
依鲁替尼(Ibrutinib)作为一种Michael受体,可选择性地与布鲁顿酪氨酸激酶(Bruton’s tyrosine kinase,BTK)的半胱氨酸残基(Cys-481)形成共价键。该激酶是至少三种关键B-细胞生存机制的重要介质。布鲁顿酪氨酸激酶的这种多重作用可以使其指挥B-细胞恶性肿瘤进行入淋巴组织,使肿瘤细胞能够接触必要的微环境而得以生存。
最初,Celera Genomics开展BTK抑制剂项目,筛选出了一系列小分子抑制剂。2006年4月,Pharmacyclics公司从Celera Genomics公司购得BTK抑制剂项目,随后选择PCI-32765(即现在的依鲁替尼)进行后续的临床前研究。2011年12月,Pharmacyclics公司与杨森制药(Janssen)合作共同开发依鲁替尼和市场推广。2013年2月,依鲁替尼被FDA授予突破性治疗药物资格,并分别于2013年11月和2014年2月批准为套细胞淋巴瘤(mantle cell lymphoma,MCL)和慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)的治疗药物。
作为Pharmacyclics的旗舰产品,依鲁替尼已经在超过40个国家被批准用于治疗血癌疾病,其中包括一种类型的白血病和一种罕见的淋巴瘤。2014年,依鲁替尼为Pharmacyclics公司贡献了5.4亿美元的全球销售收入,分析师预计,依鲁替尼的年销售收入将能在未来三年内达到30亿美元以上。2015年5月,制药公司艾伯维(Abbvie)斥资210亿美元收购依鲁替尼的生产商Pharmacyclics公司。据药渡数据检索,2015年依鲁替尼的年销售额已达14.43亿美元。
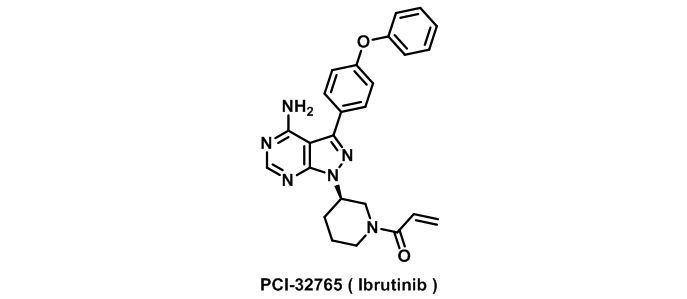
目前据文献和专利披露的依鲁替尼的合成路线主要有两条。第一条是在芳醚的结构基础上构建1H-吡唑并[3,4-d]嘧啶杂环,随后通过Mitsunobu反应,立体选择性的引入哌啶环,最后酰化,得到依鲁替尼,具体如下图所示。
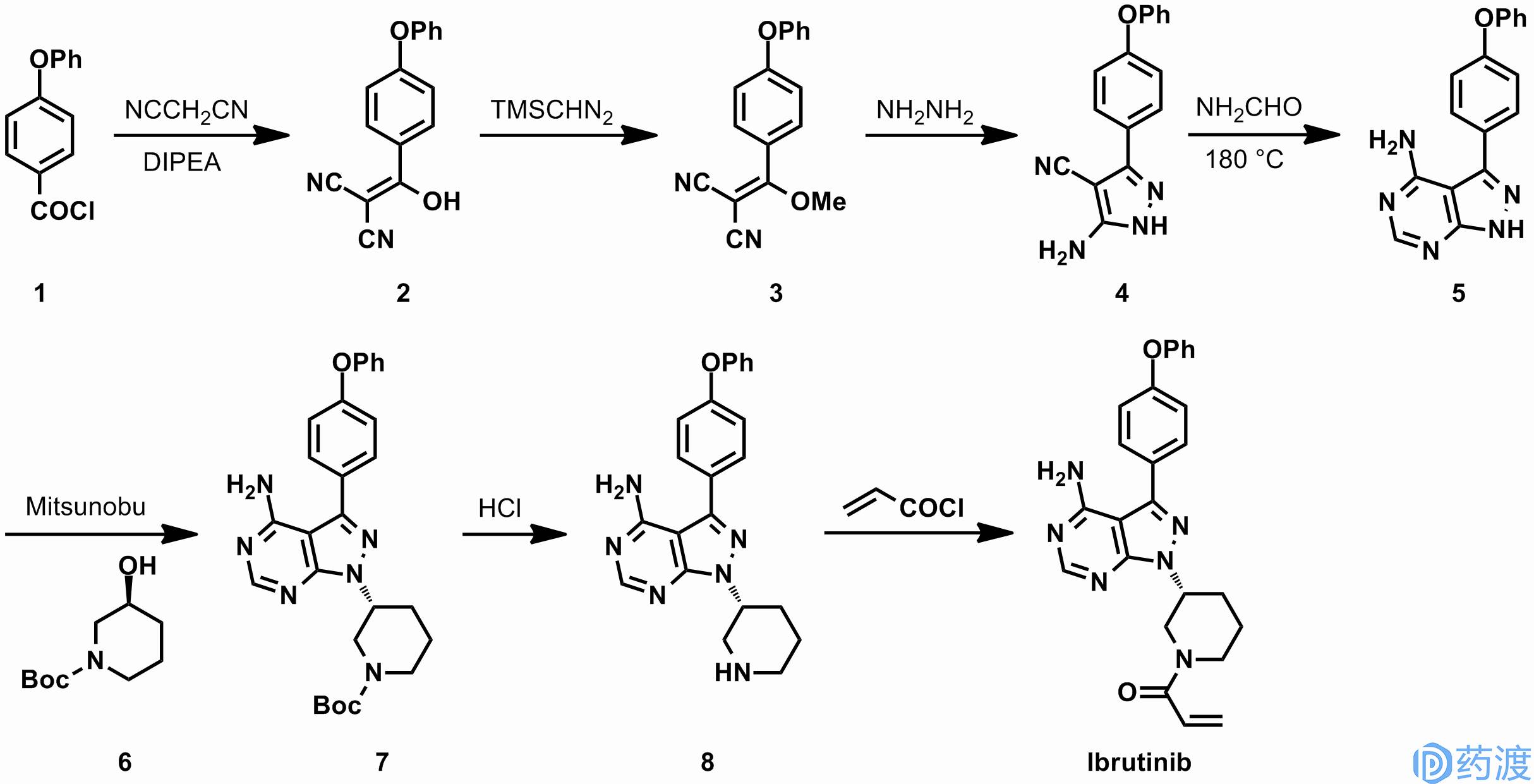
在活性筛选的早期,这条路线可以便捷的构筑不同的杂环体系,使化合物的结构多样化,有助于候选化合物的选择。之后,药物化学家对这条路线进行了简单的优化,通过对化合物
9
的碘化和Suzuki偶联,可以快速制得中间体
5
。
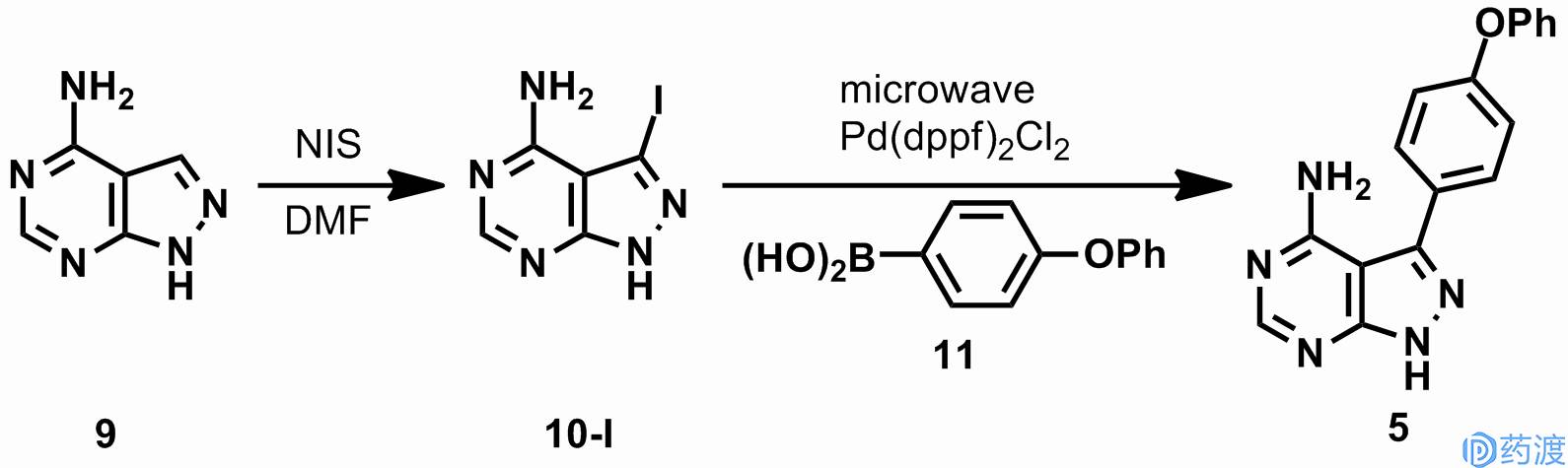
杨森制药(Janssen)申请的专利中披露了另一条生产依鲁替尼的工艺路线。化合物
3
和手性的肼
12-R
反应得到单一区域选择性的产物
13
。化合物
13
与大大过量的甲脒反应,催化氢化后得到化合物
8
粗品。粗品在甲醇与水的混合溶剂中重结晶,能以80%的收率和92.5%的纯度得到化合物
8
。
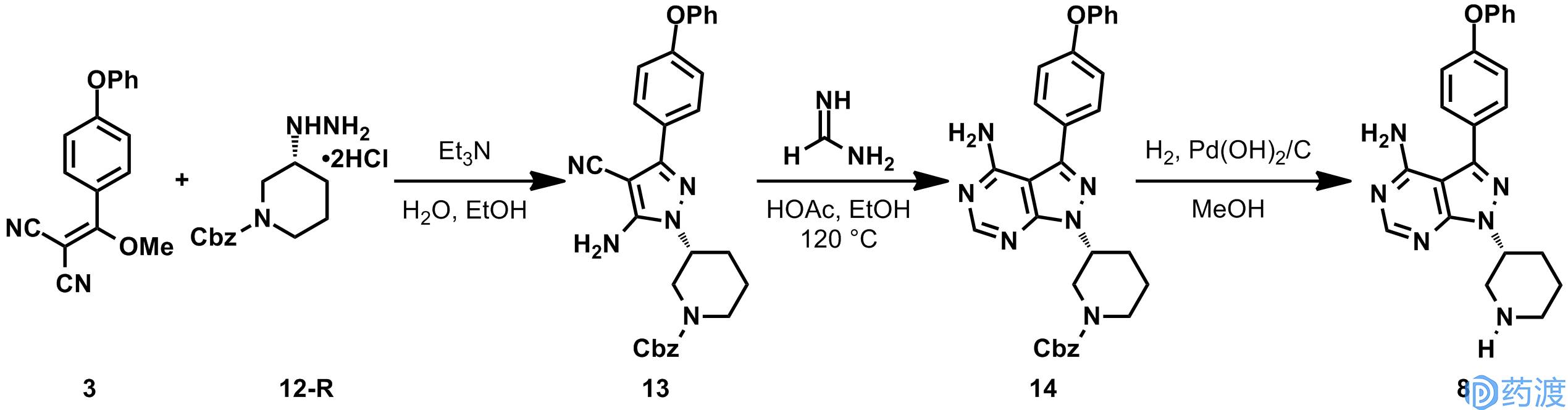
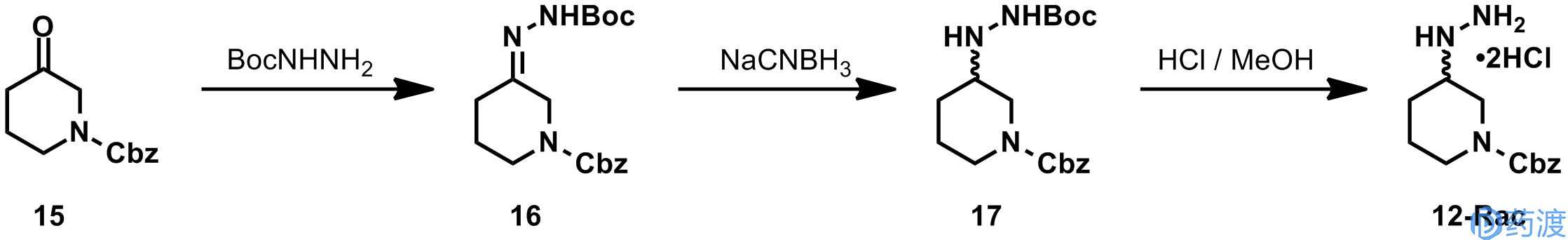
消旋的肼
12-Rac
是由化合物
15
与Boc保护的肼反应得到的腙
16
,经NaCNBH
3
还原后得到。专利中提到对
12-Rac
进行拆分,可以得到
12-R
,不过没有详细的实施例。
相较于早期的合成路线,这条制备路线具有以下优势:
①
通过化合物
12-R
,整条合成路线更具有会聚性;
②
避免使用Mitsunobu反应,减少废弃物的产生,工艺路线更加绿色环保;
③
避免使用在高温下会分解放热的致癌物质水合肼;
④
在构筑嘧啶环的步骤中,将反应温度从180℃降低到120℃,同时底物由甲酰胺替换成甲脒。
同时也有个很大的缺陷,就是要经过手性拆分才能得到手性的肼
12-R
。
鉴于其市场前景,依鲁替尼引起很多药企仿制的兴趣,不过其化合物专利最早要到2026年12月28日才会失效。目前,有众多药企开发了合成路线来制备依鲁替尼,用以规避原研的专利,主要可以分成4类。
2.1. 在工艺的最后阶段构筑吡唑并嘧啶杂环
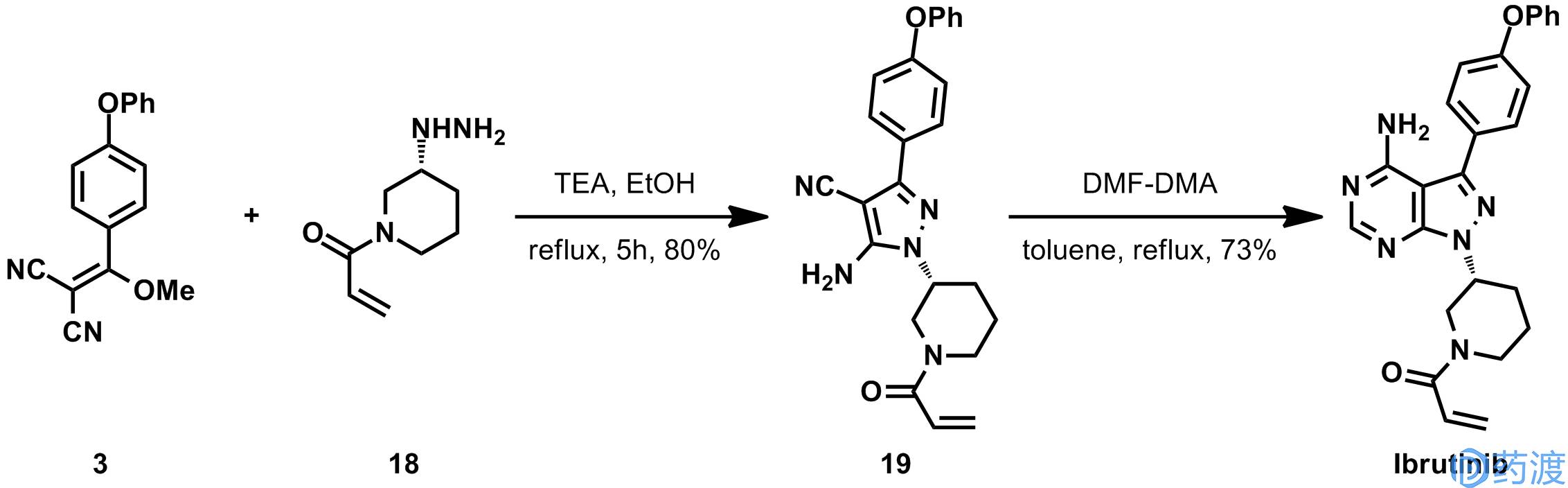
在制备的最后阶段才构筑吡唑并嘧啶杂环,化合物
3
与
18
在三乙胺的作用下形成吡唑杂环,随后与DMF-DMA反应,以73%的收率得到依鲁替尼,具体如下所示。该路线的优势就是避免了哌啶环的保护和脱保护。不过,专利中没有提化合物
18
的制备方法及其稳定性,此外化合物
18
和化合物
19
都是Michael受体,可能具有基因毒性。
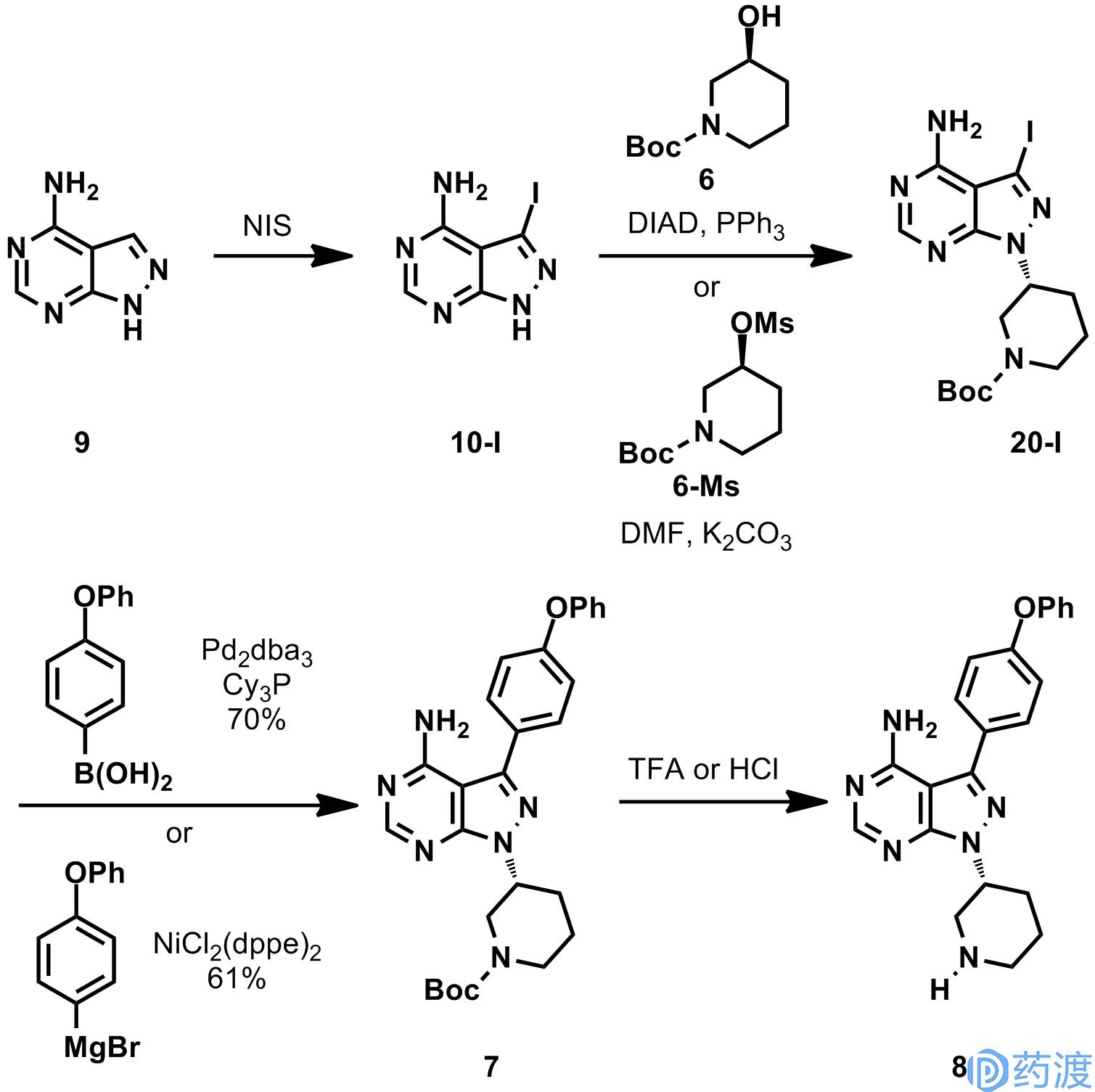
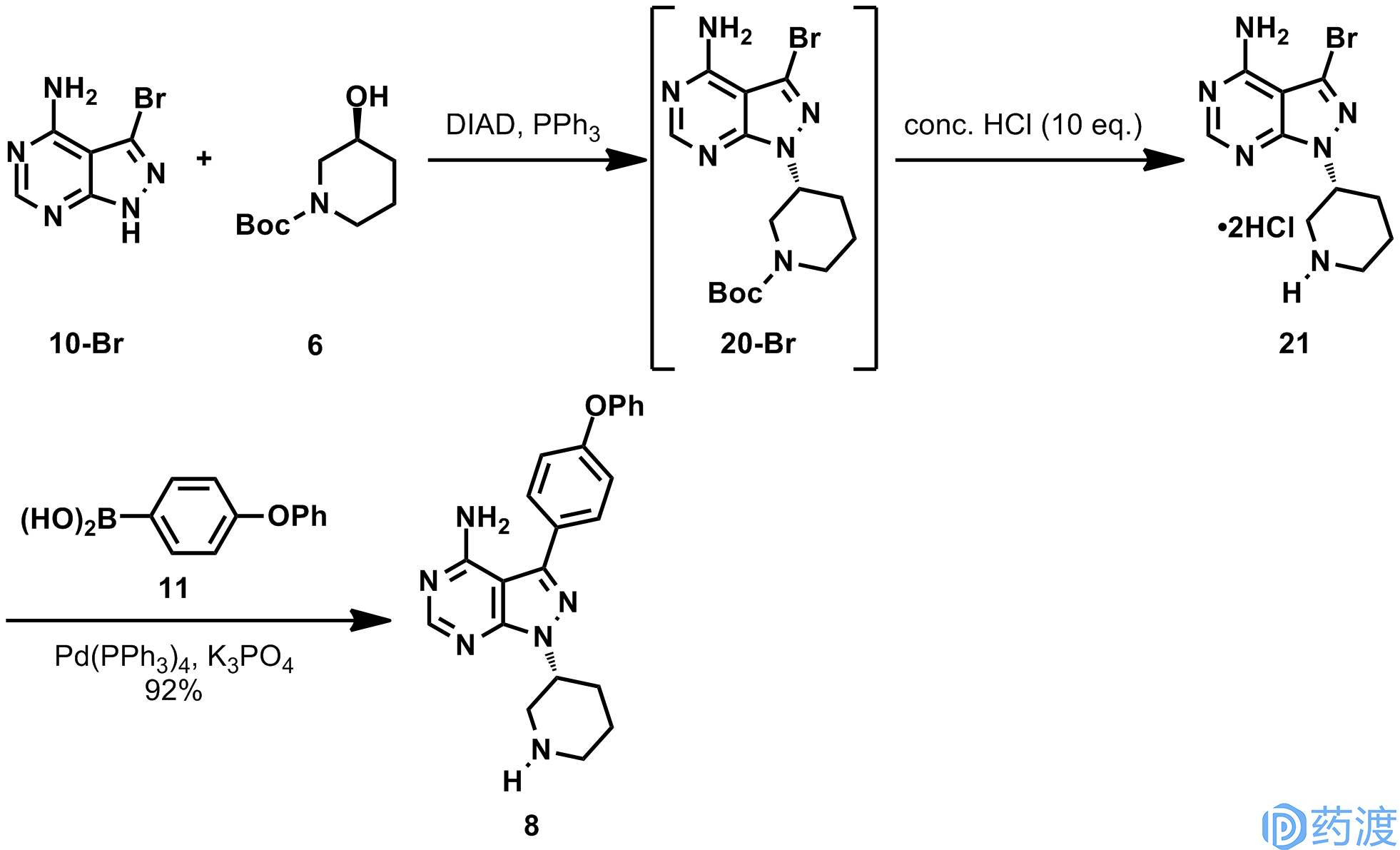
2.2. 在工艺的开始阶段先引入哌啶环,再引入芳环
在工艺的开始阶段先通过Mitsunobu反应或S
N
2引入哌啶环,再通过Suzuki或Kumada偶联引入芳环。在Arromax、Mylan、Sun Pharmaceutical和几家中国公司披露的专利中都采用类似的合成路线。化合物
9
可以大批量的商业购买,不过手性化合物
6
的生产成本较高,同时在Mitsunobu反应中,化合物
6
需要过量使用,会造成整个工艺的生产成本较高。在几篇专利中没有具体说明由此得到的依鲁替尼的纯度。
2.3. 以嘧啶环为底物构筑吡唑并嘧啶杂环
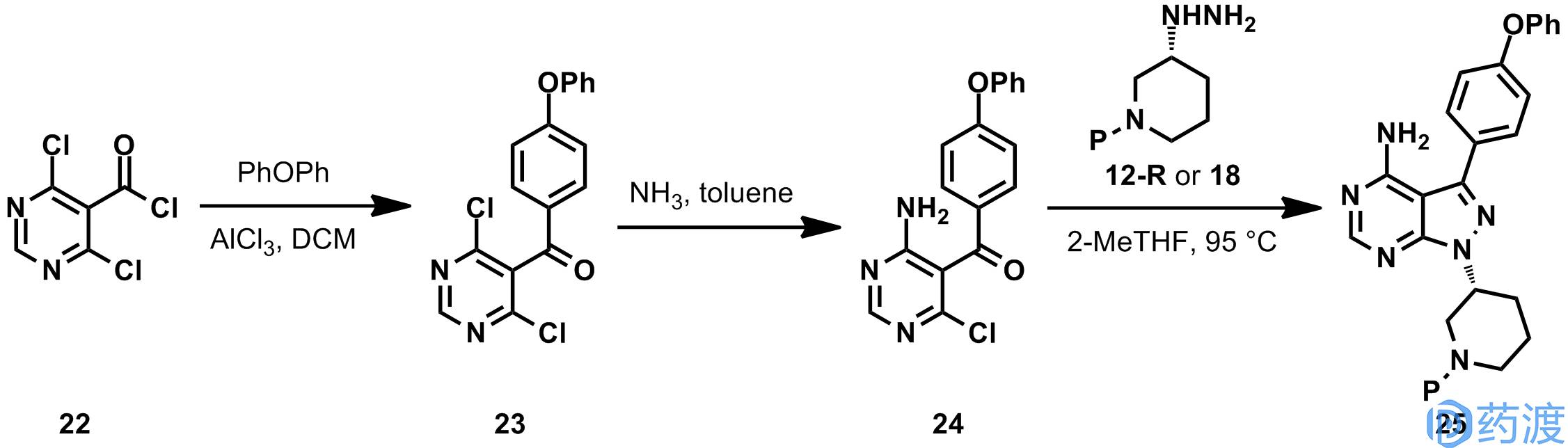
在以上提到的制备工艺中,都是在合成路线中构筑吡唑并嘧啶杂环,Sandoz和浙江台州的九洲药业通过另一种策略制得依鲁替尼。Sandoz开发的合成路如下所示,嘧啶化合物
22
与二苯醚通过Friedel-Crafts酰化反应得到化合物
23
,氨水取代,与肼反应构筑吡唑并嘧啶杂环,脱去保护得到依鲁替尼。
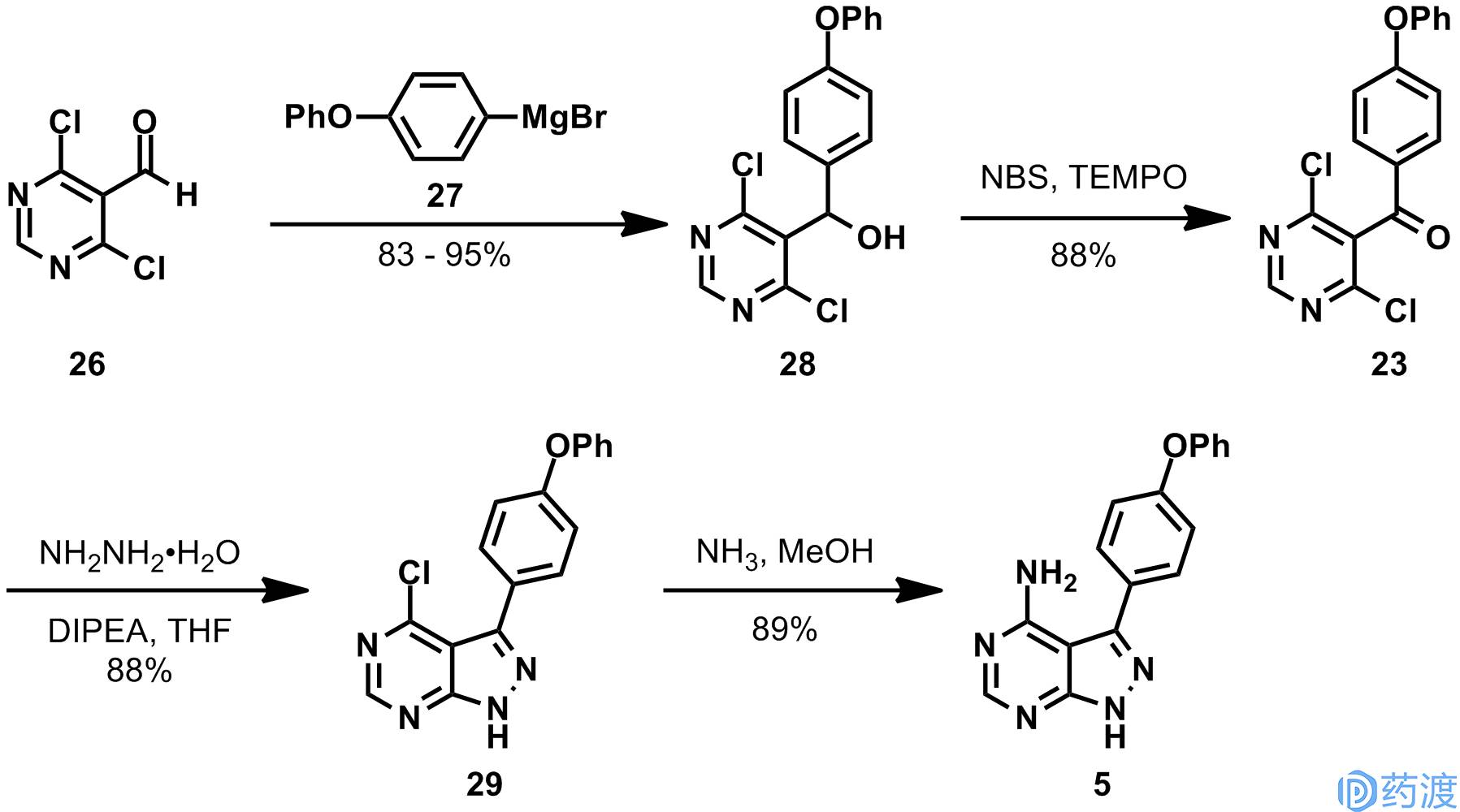
九洲药业的策略与之相类似,化合物
26
与格氏试剂得到苄醇
28
,经TEMPO/NBS氧化后,与水合肼反应构筑吡唑并嘧啶杂环
29
,氨基取代后得到重要中间体
5
。
2.4. 通过“屏蔽”丙烯酰胺得到依鲁替尼
在Sandoz披露的一篇专利中,采用了一种新颖的方式来引入依鲁替尼中的Michael受体部分。化合物
30
与化合物
5
发生S
N
2取代后,在高温下使降冰片烯部分通过逆Diels-Alder反应分解,得到依鲁替尼。这一策略的优点就是避免使用具有基因毒性的中间体,不过缺陷也很明显,反应条件过于苛刻,在最终生产放大中难以实现。






