原文:James (Wes) Barnett
在本入门教程中,我将向您展示如何创建一个盒子的水,并在恒定的温度和压力下对其进行简单模拟。 最后我们会找出水的密度。
设置
每个GROMACS模拟需要三个基本文件:结构(.gro / .pdb),拓扑(.top)和参数(.mdp)。 结构文件包含系统中每个原子位点的笛卡尔坐标。 该拓扑文件包含有关每个原子位点与其他原子位点进行联系的信息,无论该位点是处于非键联系还是键联系。 这个信息由力场提供。 非键相互作用包括范德华相互作用和库仑相互作用。 键相互作用包括键长,角度和二面角。 参数文件包括运行模拟的时间,时间步长,温度和压力耦合等信息。下面我们将获取/创建这些文件。
在这一点上,我会建议创建一个目录来存储本教程的文件。
拓扑文件
我们将从拓扑文件开始。 通常,拓扑文件使用
#include
语句来包含要使用的力场。 这个力场包括[
atomtypes
],[
bondtypes
],[
angletypes
]和[
dihedraltypes
]指令。 然后在拓扑文件中通常我们指定包含[
atoms
],[
bond
]和[
dihedrals
]的不同[
moleculetype
]指令,这些指令指向力场。 现在不要担心这个太多。 对我们来说水模型包括上述说的所有这些。有关更多信息,请参阅参考手册的第5章。
用以下文本创建一个名为
topol
.
top
的文件:
#include "oplsaa.ff/forcefield.itp"
#include "oplsaa.ff/tip4pew.itp"
[ System ]
TIP4PEW
[ Molecules ]
正如你所看到的,我们已经包含了OPLS-AA的力场。 另外我们还包括了TIP4PEW水模型。 之后你会看到一个[
System
]指令,其中只包括系统的名称,可以是任何你想要的。 最后,我们列出每种分子类型以及[
Molecules
]下的分子类型。 现在我们没有(不过马上我们将要得到这些)。
结构文件
TIP4PEW的结构已由GROMACS在拓扑目录中提供。 这个标准位置通常是
/usr/
share
/
gromacs
/
top
,但是我已经将它安装在不同的目录中。 如果您正确采购GMXRC,那么它将位于
$GMXDATA
/
top
。 在该目录中,您会看到几个
.
gro
文件,其中之一是
tip4p
.
gro
。您还会看到上面我们的拓扑文件中包含的文件夹
oplsaa
.
ff
。没有特别的
TIP4PEW
结构文件。
TIP4P
和
TIP4PEW
的四点水结构基本相同。 他们仅是力场参数不同。
要使用该结构文件创建一盒水,请执行以下操作:
$ gmx solvate -cs tip4p -o conf.gro -box 2.3 2.3 2.3 -p topol.top
如果你打开
topol
.
top
文件,你会看到最后添加了一行
SOL
和number。
SOL
是在
oplsaa
.
ff
/
tip4pew
.
itp
中定义的
moleculetype
的名称。 当我们运行
gmx solvate
时,GROMACS在每个方向2.3 nm的盒子中添加了足够的水分子。
参数文件
现在我们需要一组参数文件,以便GROMACS知道如何处理我们的起始结构。 模拟几乎总是有三个主要部分:最小化,平衡和生产。 最小化和平衡可以分解为多个步骤。 这些都需要它自己的参数文件。 在这种情况下,我们将进行两次最小化,两次平衡和一次生产运行。
我们将要使用的文件可以从这里下载。
我们所有五个文件都有一些共同点。 在每个描述中,我只给出一个非常小的注释。有关每个选项的更多信息,请参阅GROMACS页面。
|
参数
|
值
|
解释
|
|
cutoff-scheme
|
Verlet
|
用于创建邻近列表。 这是现在的默认设置,但我们在这里提供它以避免任何注释。
|
|
coulombtype
|
PME
|
使用 Particle-Mesh Ewald for long-range (k-space) 点荷联系.
|
|
rcoulomb
|
1.0
|
Cut-off for real/k-space for PME (nm).
|
|
vdwtype
|
Cut-off
|
van der Walls forces cut-off at rvdw
|
|
rvdw
|
1.0
|
Cut-off for VDW (nm).
|
|
DispCorr
|
EnerPress
|
VDW对能量和压力的长程校正。
|
应该设置截断距离,同时要记住力场是如何参数化的。换句话说,看看描述力场如何创造的期刊文章是一个好主意。我们在这里选择了1.0纳米作为截止点,这对于OPLS来说已经足够普遍了,但是您可以确定系统选择其他方法。
此外,在每个部分中,我们还将输出能量文件,日志文件和压缩轨迹文件。输出的速率(在模拟步骤中)分别使用
nstenergy
,
nstlog
和
nstxout
-
compressed
进行设置。我们将在生产运行中输出更多信息。
对于每个部分,除了第二次最小化之外,我们还将通过设置
constraint
-
algorithm
=
lincs
和
constraints
=
h
-
bonds
,使用LINCS算法约束所有涉及氢的键。这使我们能够使用比其他更大的时间步骤。
对于第一次最小化,我们使用最陡峭下降算法,通过设置
integrator
=
steep
来最小化系统能量,最大步长为1000步(
nsteps
=
1000
)。如果在此之前能量收敛,则最小化将停止。另外我们进行
define
=
-
DFLEXIBLE
。这让GROMACS知道使用灵活的水,因为默认情况下所有的水模型都是使用
SETTLE
算法为刚性模型。在我们拥有的水模型的拓扑文件中,有一个if语句查找要定义的
FLEXIBLE
变量。第一次最小化的目的是使分子处于良好的起始位置,这样我们就可以无任何错误地打开
SETTLE
。
在第二个最小化中,我们只需删除
define
=
-
DFLEXIBLE
并将最大步数增加到50,000。
最后三部分-两个平衡和生产-都使用
integrator
=
md
。此外,通过设置
dt
=
0.002
来使用2fs时间步长。
对于第一个平衡步骤,有几点需要注意。我们正在添加如下所示的几个参数:
|
参数
|
值
|
解释
|
|
gen-vel
|
yes
|
根据Maxwell-Boltzmann分布为每个原子位点生成速度。
只为您的第一个平衡步骤生成速度
。这使我们接近我们将耦合系统的温度。
|
|
gen-temp
|
298.15
|
K中的温度用于
gen
-
vel
。 除非你正在做一些奇怪的/有趣的事情,否则这应该与ref-t相同。
|
|
tcoupl
|
Nose-Hoover
|
用于温度耦合的算法。 Nose-Hoover 为经典温度耦合算法。
|
|
tc-grps
|
System
|
要结合哪些组。 你可以将不同的原子组分开,但我们只需要耦合整个系统。
|
|
tau-t
|
2.0
|
耦合的时间常数。 详情请参阅手册。
|
|
ref-t
|
298.15
|
以K为单位的温度。
|
|
nhchainlength
|
1
|
Leap-frog integrator 只支持1,但默认情况下这是10。这是设置,所以GROMACS不会发出警告。
|
第一次平衡的关键是在加压耦合之前让我们达到正确的温度(298.15 K)。 同时添加温度和压力耦合可能会导致系统不稳定并发生崩溃。 我们不想在一开始就震惊我们的系统。 另外,我们设置了
nsteps
=
50000
,所以以2fs的时间步长,这意味着它将运行100ps。 这对我们在这里所做的工作是足够的,但是在更大/更复杂的系统中,您可能需要更长时间的平衡。
第二个平衡增加了压力耦合。 请注意,我们并没有再次产生速度,因为那样会取消我们刚刚做的一些工作。 我们还为约束设置了
continuation
=
yes
,因为我们从第一次平衡开始继续进行模拟。 这部分将运行1ns。 同样,对于其他系统,这可能需要更长一些。
|
参数
|
值
|
解释
|
|
pcoupl
|
Parrinello-Rahman
|
用于压力耦合的算法。 Parrinello-Rahman在与Nose-Hoover一起使用时正确地产生了等压等温线。
|
|
tau-p
|
2.0
|
耦合的时间常数。 详情请参阅手册。
|
|
ref-p
|
1.0
|
压力耦合常数
|
|
compressibility
|
4.46e-5
|
系统压缩系数
|
|
对于生产运行,除了输出更多数据并运行10 ns外,所有内容与上次平衡完全相同。
|
|
|
模拟
我们现在有了我们需要的所有文件来运行模拟的每个部分。 每个部分通常运行
gmx grompp
,以将我们现在具有的三个文件(.gro,.top和.mdp)预处理为.tpr文件(有时也称为拓扑文件)。
最小化
首先,通过执行以下操作来执行我们的两个最小化步骤:
$ gmx grompp -f mdp/min.mdp -o min -pp min -po min
$ gmx mdrun -
deffnm min
$ gmx grompp -f mdp/min2.mdp -o min2 -pp min2 -po min2 -c min -t min
$ gmx mdrun -deffnm min2
在每个部分,我们都使用
-
f
标志在.mdp文件中读取。默认情况下,如果未指定
-
c
和
-
p
标志,GROMACS将使用
conf
.
gro
和
topol
.
top
作为结构和拓扑文件。此外,我们正在输出处理后的拓扑文件
-
pp
和mdp文件
-
po
。这些是可选的,但可能值得一看,尤其是处理过的mdp文件,因为它已被评论。
在接下来的每一步中,我们使用
-
c
和
-
t
标志读入前一步的最后一个结构文件或检查点文件。默认情况下,GROMACS每15分钟和最后一步输出检查点文件。如果检查点文件不存在,GROMACS将使用由
-
c
定义的结构文件,因此指定两者都是一种很好的做法。在每个
gmx mdrun
中,我们都告诉GROMACS为每个输入和输出文件使用默认名称,因为会输出多个文件。
注意我们使用
-
maxwarn
1
来进行第二次最小化。只有使用这个标志,如果你知道你在做什么!在这种情况下,我们会收到关于我们可以安全绕过的L-BFGS效率的警告。
为了了解发生了什么,让我们使用GROMACS命令
gmx energy
来提取这两个部分的势能。执行以下操作并输入与
Potential
对应的数字,然后再次输入:
$ gmx energy -
f min.edr -o min-energy.xvg
相似的行为进行第二次能量最小化的阅读
$ gmx energy -f min2.edr -o min2-energy.xvg
结果
.
xvg
的标题。 文件将包含供Grace绘图程序使用的信息。 我使用gnuplot,所以这些行中的一些会导致错误。 我用
.
xvg
中的
#
替换每个
@
字符。然后我可以使用gnuplot。首先启动gnuplot进行绘图:
$ gnuplot
在gnuplot终端输入如下命令:
> plot 'min-energy.xvg' w l
第二次如下:
> plot 'min2-energy.xvg' w l
第一次的结果类似如下:
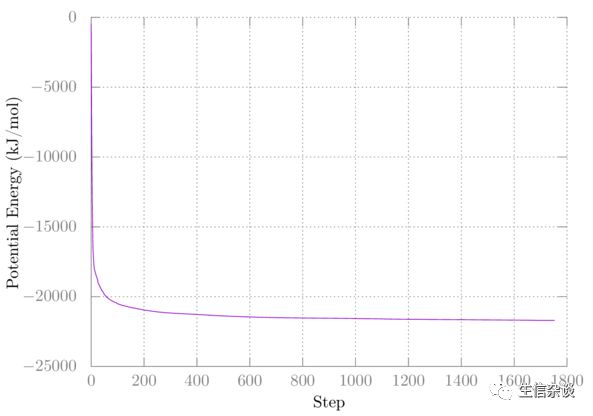
第二次结果没有改变,所以未在此绘出
Equilibration 1 (NVT)
现在我们有了一个好的起始结构,让我们通过添加温度耦合来完成第一个平衡步骤:
$ gmx grompp -f mdp/eql.mdp -o eql -pp eql -po eql -c min2 -t min2
$ gmx mdrun -deffnm eql
我们来看看整个模拟过程中温度如何变化:
$ gmx energy
-f eql.edr -o eql-temp.xvg
在提示符下选择与温度相对应的数字,然后再次输入。 像上面那样在gnuplot中绘制它。 你应该看到像这样的东西
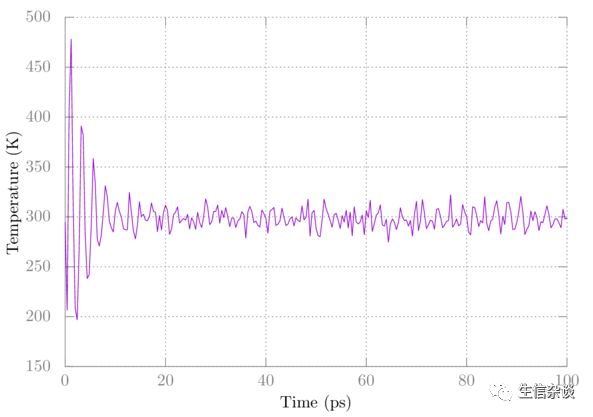
请注意,温度最初会波动很大,但最终会稳定下来。
Equilibration 2 (NPT)
如前所述,对于我们上次的平衡,我们添加了一个压力耦合:
$ gmx grompp -f mdp/eql2.mdp -o eql2 -pp eql2 -po eql2 -c eql -t eql
$ gmx mdrun -deffnm eql2
您可以使用上述
gmx energy
检查温度和压力。绘图类似如下
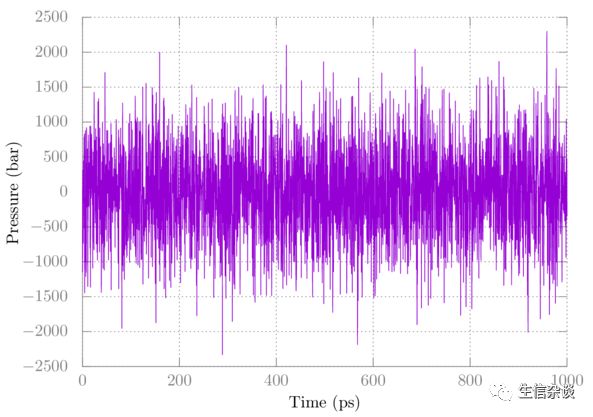
请注意,压力波动很大,这是正常的。 在这种情况下,完全平衡后的平均值应接近1 bar。
正式模拟
正式模拟部分如下:
$ gmx grompp -f mdp/prd.mdp -o prd -pp prd -po prd -c eql2 -t eql2
$ gmx mdrun -deffnm prd
分析
在
prd
.
edr
上使用上面的
gmx energy
,得到平均温度,压力和密度。 他们是你期望的吗?
这是我的输出:
Energy Average
Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Temperature 298.145 0.019 8.65629 0.0338992 (K)
Pressure 3.25876 0.97 688.616 -2.75083 (bar)
Density 995.381 0.15 12.92 0.0705576 (kg/m^3)
如果你看图4中的TIP4PEW论文,你可以看到我们已经达到了正确的密度。 另外请注意,Wolfram Alpha表示标准条件下的水密度为997 kg /立方米。
你也可以使用像vmd这样的程序来模拟你的模拟。 用vmd打开正式模拟的轨迹:
$ vmd prd.gro prd.xtc
快照如下:
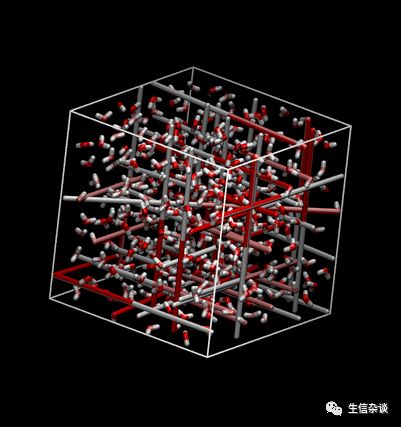
进行周期性处理
$ gmx trjconv -f prd.xtc -s prd.tpr -pbc mol -o prd-mol.xtc
后结果如下:

总结
在本教程中,我们使用
gmx solvate
物产生一盒TIP4PEW水。 我们在五个不同的部分对它进行了模拟:最小化1,最小化2,平衡1,平衡2和生产。 每个部分都使用自己的.mdp文件进行了解释。 在每个部分,我们使用
gmx energy
来提取有关模拟的有用信息。 生产运行后,我们能够找到TIP4PEW水的密度。
问题
如果教程中存在错误或错误,或者如果有什么不清楚的地方,请打开一个问题,我很乐意解决它。 如果您有关于GROMACS的一般问题,请参阅GROMACS文档。 您还可以将关于GROMACS的一般问题通过查阅邮件列表来解决。





