正文
投稿:热心小网友
校正:
AspirinCode_W
同源模建
基因复制和趋异同化产生了同源蛋白族系,同源的序列可能来自共同的祖先。蛋白质的同源性使人们有可能根据己知的蛋白质三维结构去推测未知结构的同源蛋白质的初步结构,人们通过对类似蛋白质空间结构的对比发现,蛋白质的三维结构比蛋白质的一级结构更加保守,而后者又比
DNA
序列更为保守。因此,用类似物的三维结构来预测蛋白质结构是比较可靠的。
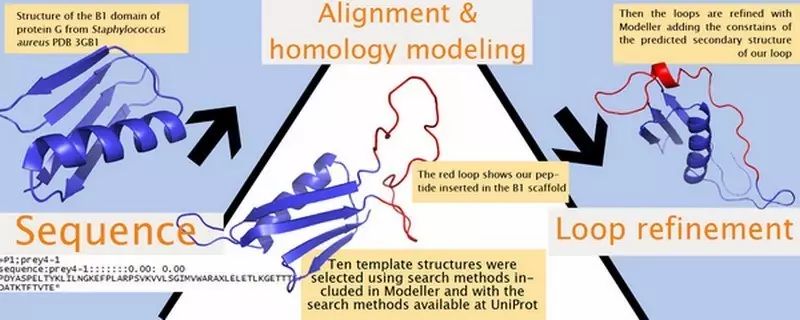
生物进化过程中,有时
DNA
序列变异而会引起蛋白质的氨基酸序列突变,可是维持蛋白质功能和结构部分的氨基酸序列不会变化。一般说来,负责维持功能的氨基酸常位于蛋白质的表面,而负责维持结构的氨基酸多位于蛋白质的核心,在进化过程中有较大变异的氨基酸则多在环状结构
(loop)
上,将氨基酸序列和结构进行比较,如果两个序列的一致性
(identity)
超过
40%
,则此两种蛋白质就判定为同源蛋白。同源模建的基本假设是序列的同源性决定了三维结构的同源性,一个未知结构的蛋白质分子
(
目标蛋白
)
的结构是可以通过与之序列同源且结构已知的蛋白质
(
模板蛋白
)
来进行预测的。
同源建模的一般步骤
(1)
同源蛋白的搜索
在搭建目标蛋白模型之前,首先需找出与目标蛋白序列同源且已知晶体结构的模板蛋白。常用来进行同源蛋白搜索的程序有
FASTA
和
BLAST,
其中
FASTA
是第一个广泛使用的数据库相似性搜索程序。该程序是引用代矩阵实行局部比对以获得最佳搜索。因此本实验采用
FASTA
程序进行搜搜。
(2)
序列比对
序列比对和排列是同源模建的关键步骤,它通过对目标蛋白与模板蛋白的序列进行排列和定位可确定出序列的保守区域。

序列比对是通过一系列得分矩阵实现的。常用的得分矩阵有同源性矩阵(
identity matrix
)、密码子矩阵(
codon substitution matrix
)
、突变矩阵(
mutation matrix
)、疏水性矩阵(
hydrobocity matrix
)
等。比对程序在比对过程中,将其中一个序列看作行,将另一个序列看作为列,进而构造出上面所提到的几种矩阵。通过对矩阵内的每一个残基单位进行比对,进而得到得分最高的排列方式。常用的序列比对程序有
EMBL-EBL
,这个程序都只需在网上提交模板序列与目标序列即可运行。
(3)
模型搭建
模型搭建主要包括给保守区和柔性的序列区赋坐标。当目标蛋白和模当目标蛋白和模板蛋白完成序列比对以后,就可给保守区的氨基酸残基赋坐标。若两者的序列完全相同,则把模板蛋白相应的残基坐标直接拷贝给目标蛋白的残基即可;若两者的残基不同,则把模板蛋白主链坐标拷贝给目标蛋白,对于侧链则尽量保持其相似的结构模式;若目标蛋白的的侧链大于模板蛋白,则无法匹配的构象取其扩展构象。保守区以外的区域则为非保守区,非保守区常具有一定的柔性,因此又称为柔性区。在模型搭建过程中,目标蛋白柔性区的搭建主要有两种方法:一种是数据库搜索法;另一种为随机产生法。因目标蛋白柔性区的建立是基于统计法与随机搜索法,所以会引入一定的误差。本实验同源模建采用的软件是:
SWISS-MODEL
。
(4)
模型优化
模型产生后需对分子结构进行进一步优化。优化的目的是用来消除原子间的重叠以及不合理的构象,尤其是柔性区的构象。优化一般采用分子力学与分子动力学的方法。
(5)
模型评价
用来评价模型的指标主要包括立体化学和能量评价两个方面。本实验立体化学的评价采用
PROCHECK
程序来执行的,模型的能量可用
PROSA
程序执行。
PROCHECK
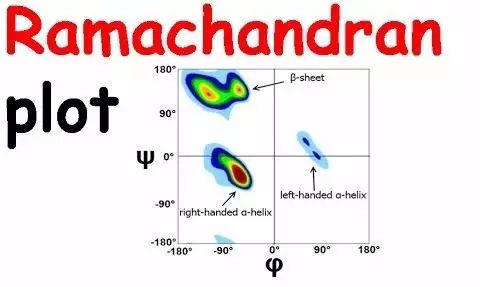
主要是评价模型中残基与残基之间的立体化学性质,考察的是残基之间的
φ
和
ψ
两个角度分布在
Ramachandran
图中的分布是否合理。
PROSA
程序是模型能量方面的评价。用于评价每个残基之间的相互作用能是否在一个合理的范围内。
同源模建法是唯一比较快速、精确的预测蛋白结构的可靠方法,在预测蛋白三维结构方面,比目前实验手段所能达到的数量和速度要快十倍以上,具有准确性最高、发展最成熟的优点,所以,对于一般药物设计来说,主要采用同源模建的方法。通过同源模建,能够很方便地建立未知蛋白的结构模型。因此,本实验决定采用同源模建的方法对靶点蛋白进行模建,为后续的实验研究结果作出合理解释并为实验提供指导性的信息。
分子对接
分子对接是受体空腔与配体小分子进行相互识别的过程,他们的识别过程遵循几何能量匹配和化学环境互补的原则,首先通过搜寻受体分子结构的活性位点,将小分子置于活性口袋中,然后分子对接程序计算活性中心的各个方位,打分函数搜寻小分子的最佳结合方式(空间构象最优,空间位置最优)。受体分子与配体结合的部位是蛋白质的某几个氨基酸残基,结合的方式包括静电作用、氢键作用、范德华相互作用和疏水作用。其中静电作用、氢键作用和范德华力控制药物
-
受体结合,疏水作用是药物
-
受体结合的驱动力,如果疏水作用强,药物和受体就能排除水分子,相互结合在一起。分子对接在药物设计中有重要意义
。
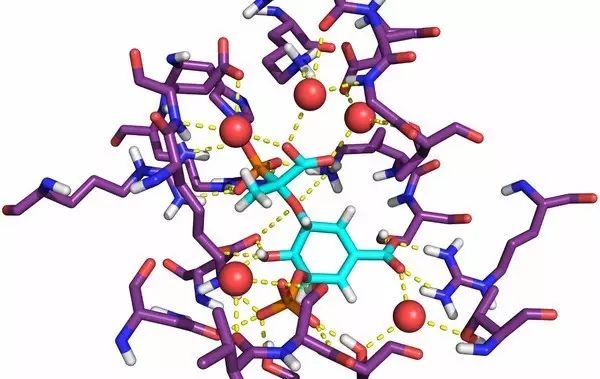
1.
范德华力
:存在于分子间的一种吸引力,它比化学键弱得多。
包括色散力、诱导力和取向力。
2.
氢键
:氢键是一个活性氢原子与两个电负性原子形成的键,氢键中两电负性原子的距离一般为
2.5~2.7Å
,不超过
3Å
,虽然氢键的强度只是共价键的
1/10
,但是氢键作为药物分子和受体表面结合的键能之一,它能影响药物的物理性质以及影响生物活性。
3.
疏水性相互作用
:在蛋白质分子中有缬氨酸(
Val
)、亮氨酸(
Leu
)、异亮氨酸(
Ile
)等氨基酸单位的存在,它们具有较大的烷基,具有建立疏水性键的能力。这种能力有两个方式表现:
a.
其在高分子量蛋白质的大而易弯曲的分子中形成分子内疏水性键,这种键可促进和稳定键的折叠和造成疏水性小袋或裂痕,从而形成蛋白质的四级结构。
b.
药物分子的疏水区和天然大分子的疏水区之间的相互作用在药物
-
大分子复合物的形成和稳定中起重要作用,这种相互作用是药物
-
蛋白质结合的突出特点。
4.
静电相互作用
:
a.
两相反电荷的离子相互作用(离子键):在生理p
H
下,肽和蛋白质的赖氨酸(
Lys
)和精氨酸(
Arg
)末端碱基以及天冬氨酸和谷氨酸单位的游离羧酸全部电离产生阳离子和阴离子中心,它们能与离子化的药物分子形成离子键,从而产生作用。具有较弱碱性和酸性中心的组氨酸(
His
)和酪氨酸(
Tyr
)等也包括在内,但程度较低。
b.
一个离子与一个分子偶极的相互作用。
c.
两个分子的偶极相互作用。
d.
一个离子与一个诱导偶极的相互作用。
分子对接包括钢性对接,半柔性对接,柔性对接
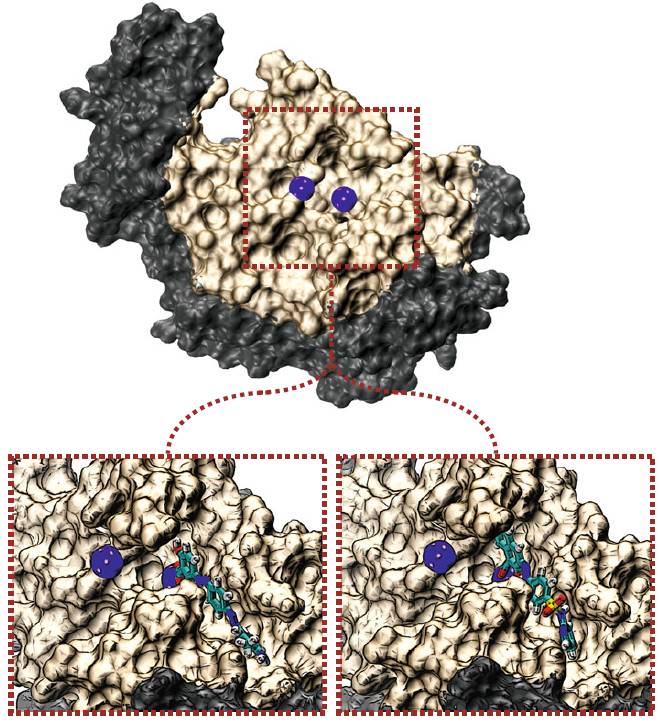
(1)
刚体对接
在对接过程中,研究体系
(
主体与客体
)
的构象不发生变化。适合考察结构比较大的体系,如蛋白质和蛋白质间以及蛋白质和核酸等大分子间的对接。
Stoddard
等
用二元对接方法成功地对麦芽糖和蛋白质进行了对接。对接时,他们把配体和受体主链结构当作刚性处理。
(2)
半柔性对接
在对接过程中,研究体系尤其指配体的构象允许在一定的范围内变化,但大分子是刚性的
,
构象不变化。适合处理大分子和小分子间的对接,
因为受体大分子构象的变化相对底物分子而言变化不大。比如蛋白质分子与配体小分子的作用
。进行半柔性对接时,一般使用快速的对接软件
-FIexX
,因为它允许配体是柔性,而受体是刚性的。
(3)
柔性对接
在对接过程中,底物和受体的构象是允许发生变化的。一般用于精确考虑分子间的识别情况。分子的柔性主要来自于可旋转键的旋转。这种变化包括三个平动自由度、三个转动自由度以及底物分子的部分二面角的变化,
Mangoni
等用柔性的配体对柔性的受体进行了对接。
分子对接方法
(Molecular Docking Method)
是药物设计方法中比较成熟的直接药物设计方法。所谓的分子对接(
molecular docking
),就是受体活性空腔与配体小分子之间通过几何、能量以及化学环境匹配而相互识别的过程。分子对接在药物设计中具有十分重要的意义。药物分子产生药效的过程,就是靶酶和药物分子充分结合作用的过程,该过程需要靶酶和药物分子采取合适的空间取向在必要的部位相互契合,并相互作用,通过构象调节得到稳定的复合物构象。受体活性空腔与配体小分子的空间匹配是分子间发生相互作用的基础
,
能量匹配是分子间保持稳定结合的基础。
(感谢网友的投稿)

本文系《药设之道》原创,
欢迎分享到个人朋友圈,
转载需授权!
投稿荐稿
:
《药设之道》(微信号:DrugDesigner)致力于传播药物设计
新技术、新方法、新理论、新观点和最新的药物研究动态
,是专注于药物设计的信息分享平台,投稿和荐稿请联系:[email protected]





