作者简介: 入行不深,3年有余。精雕技术,广不足而深有余。机器学习,好之适之点缀生信。 人称黑马王子,凡有涉猎,必得口令,便至臻境。 关于创作,外号“三不先生”:书上有的不写,别人写过的不写,自己写过的不重写。 篇篇原创,与君共赏。 |

VarScan2概述
VarScan2是华盛顿大学开发的一款多用途的call变异工具。可用于种系细胞、体细胞(SNV/INDEL/CNV/)(CNV不建议用,据说是别人提供的脚本,漏洞百出)、de Novo Mutations(trios))等。
VarScan2使用各种过滤方法,以得到各位点在样品中的基因型,并计算各等位基因的read 频率。此工具只支持两种等位基因的基因型。通过比较两个样品在该位点各等位基因的read 频率,并通过fisher’s检验得到差异显著性水平p 值。VarScan2工具将突变位点分为三种突变状态:somatic 突变、germline 突变、LOH 突变;并从三种状态的位点,得到置信位点。
以肿瘤对照样本为例,做下具体说明(推荐VarScan2,不要用varscan):
mpileup文件生成
因为varscan使用pileup作为输入格式,我们需要先将bam处理为pileup格式,有2种方法
方法1:生成2个pileup文件
samtools mpileup -q 1 -d 30000 -L 30000 -f ucsc.hg19.fasta normal.bam > normal.pileup
samtools mpileup -q 1 -d 30000 -L 30000 -f ucsc.hg19.fasta tumor.bam > tumor.pileup
方法2:生成1个mpileup文件(官方文档强烈推荐)
samtools mpileup -q 1 -d 30000 -L 30000 -f ucsc.hg19.fasta normal.bamtumor.bam > normal.tumor.mpileup
参数说明:-q 1表示跳过比对的mapQ得分<1的碱基
-d 30000表示bam文件的最大测序深度为30000,这是考虑超深度测序的设置
-L 30000表示INDEL序列的最大测序深度为30000,这是考虑超深度测序的设置
备注:本人试过2种方法,方法1在后续的肿瘤变异calling时总是报错,是不是现在不能用了?建议用方法2。
肿瘤配对样本分析
Java -jar VarScan.v2.4.3.jar somatic normal.tumor.mpileup normal.tumor.vcf --mpileup 1 --output-vcf 1
参数说明:
--mpileup1表示采用mpileup文件
--output-vcf1 表示输出文件为vcf格式
结果文件:这里会生成normal.tumor.vcf.snp和normal.tumor.vcf.indel文件
靶向测序区域过滤:limit命令(许多人不知道该命令!拿去不谢!)
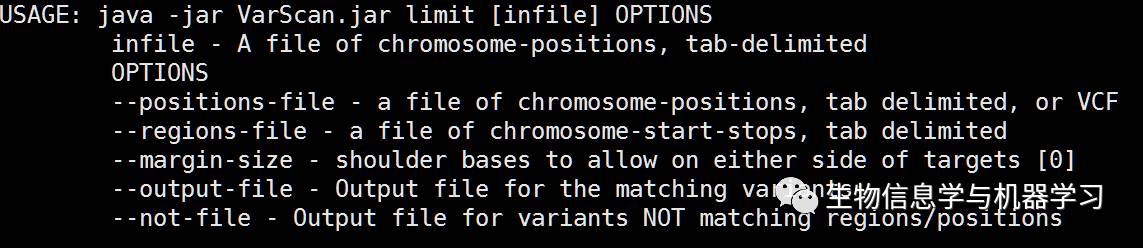
Java -jar VarScan.v2.4.3.jar limit normal.tumor.vcf.snp --regions-file regions.bed --output-file normal.tumor.vcf.target.snp
参数说明:--regions-file指定靶向测序分析的范围
变异过滤:processSomatic命令
Java -jar VarScan.v2.4.3.jar processSomatic normal.tumor.vcf.target.snp --min-tumor-freq 0.05 --max-normal-freq 0.15--p-value 0.07
参数说明:这里测序深度较深,所以--max-normal-freq0.15设置较大。
点评:
VarScan2在国内临床分析领域用得还是相当广的,不知其故?难道是变异结果文件分类很细的原因吗?如果只是因为方便而忽略准确性,我就只能呵呵了。
关于三大肿瘤工具的对比分析,请看下篇。










