FDA 20210304 培训-1
杂质确认考量---ICH Q3A/Q3C/Q3D,RLD&MDD


缩写
-
RLD---参比制剂
-
MDD---最大日剂量
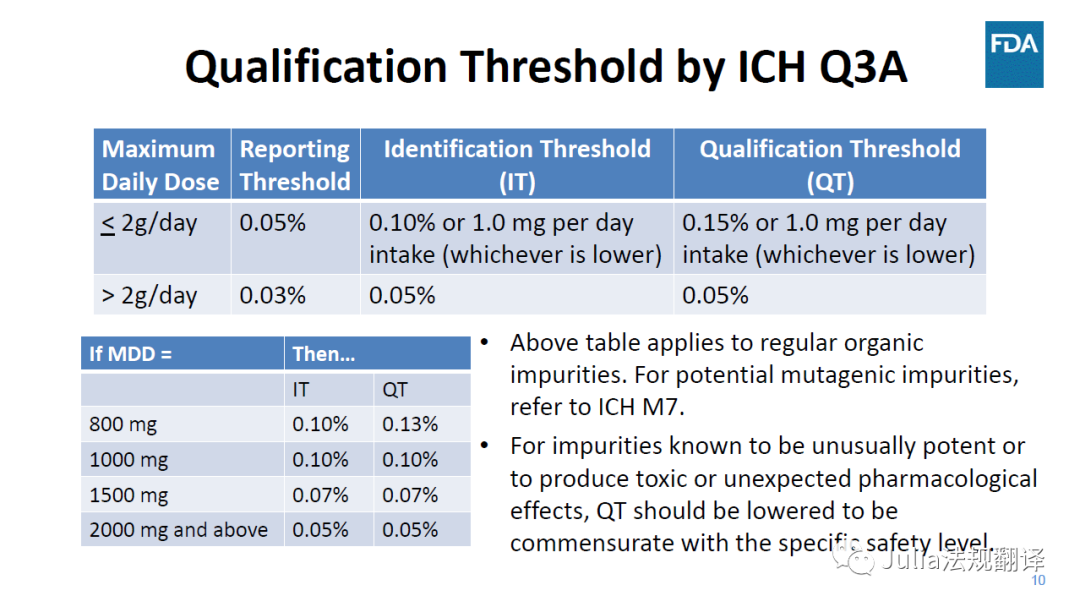
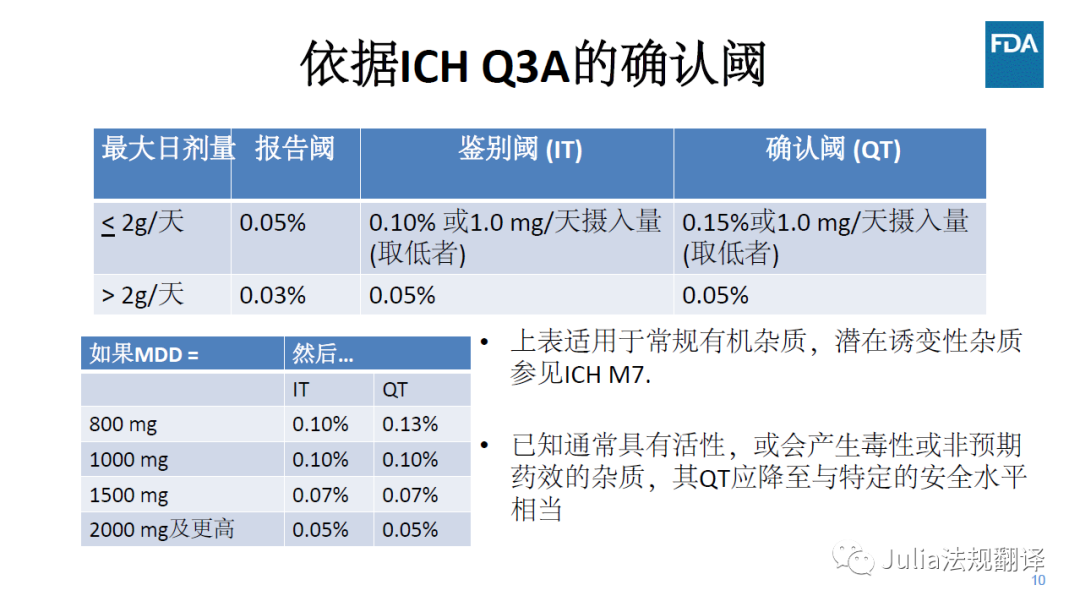
-
IT/QT---鉴别阈/确认阈
-
PDE---允许日暴露值
-
(Q)SAR---(定量)构效关键
-
Pharm/Tox---药学/毒性

提纲
-
简介
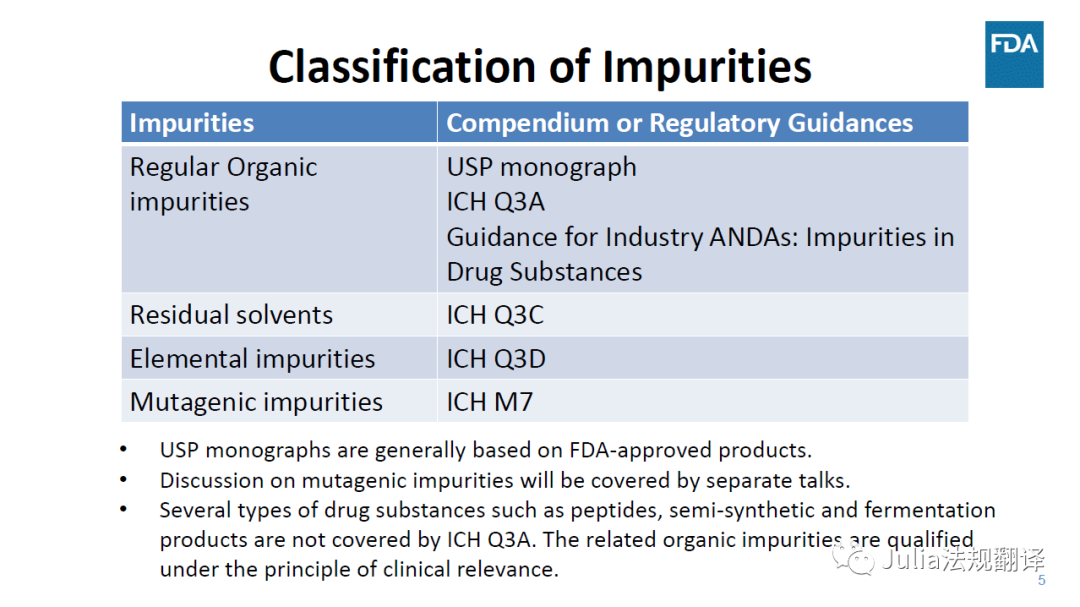
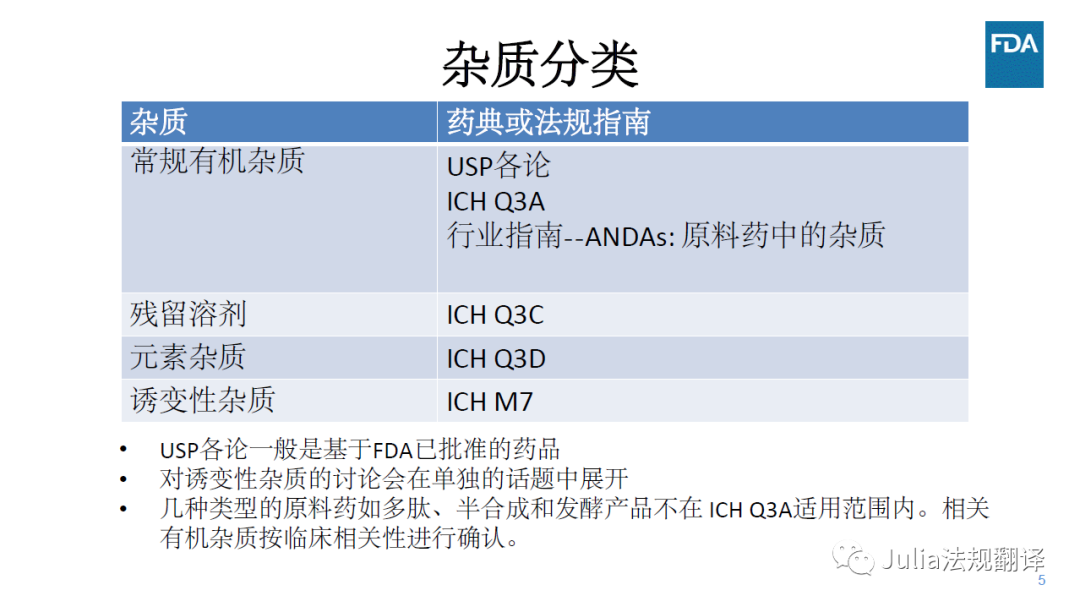
一 杂质分类和适用指南
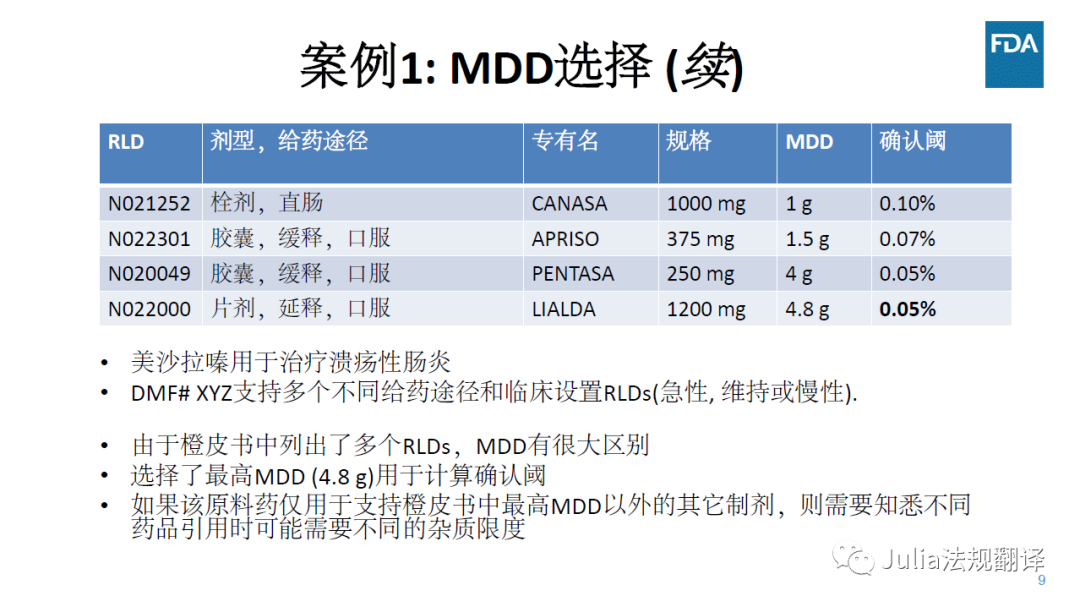
一 案例1:MDD选择
-
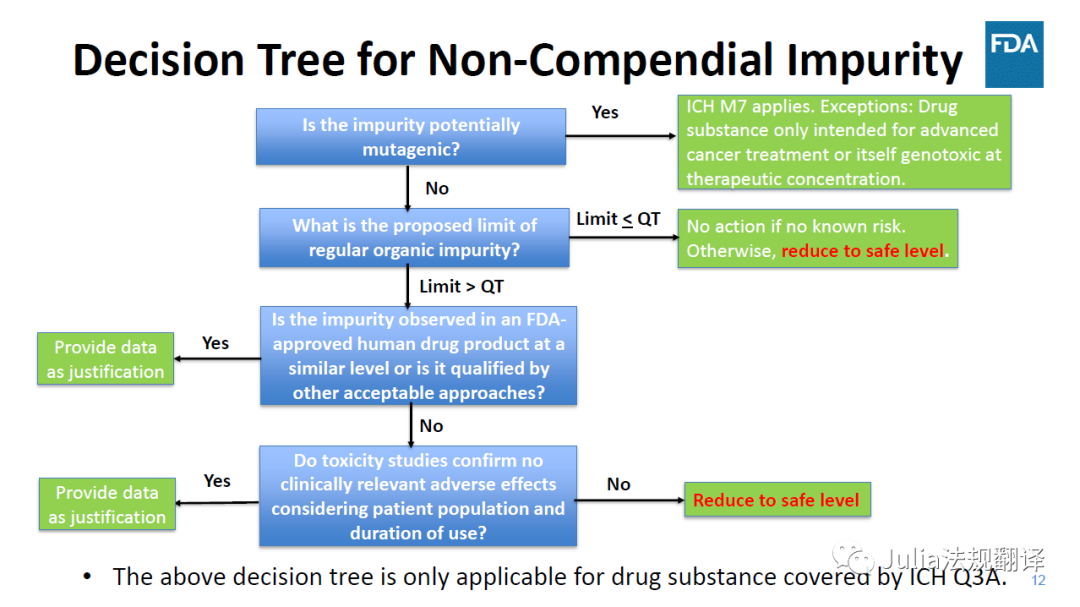
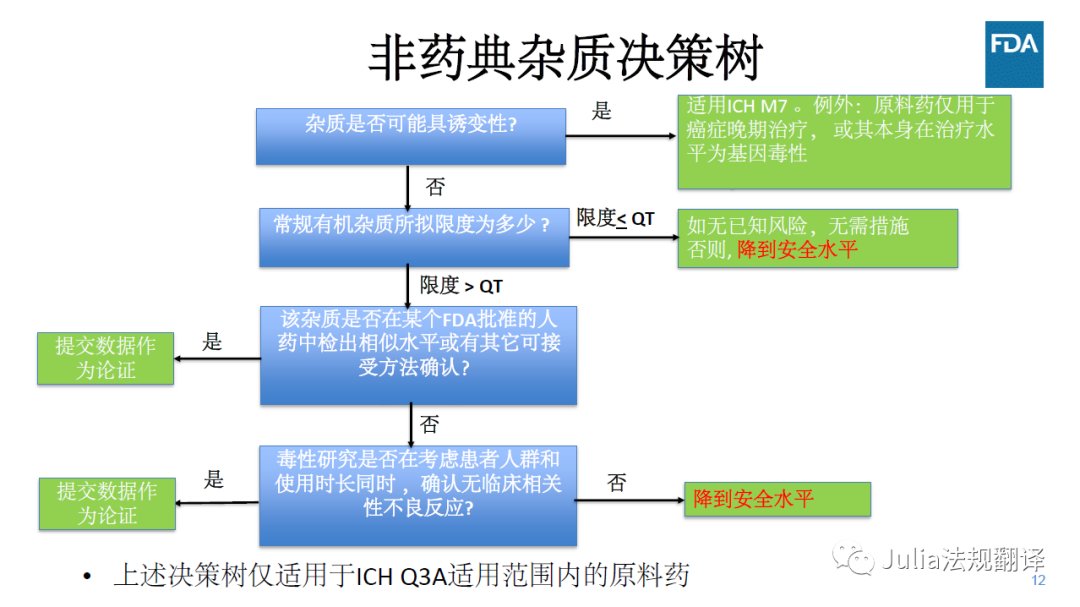
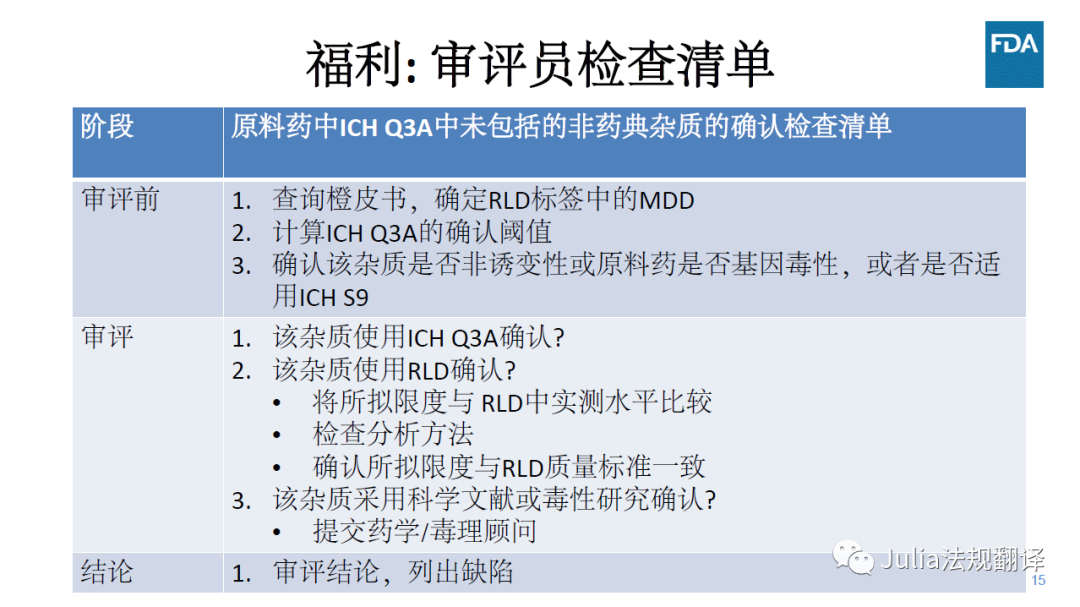
常规杂质的确认
一 决策树和确认方法
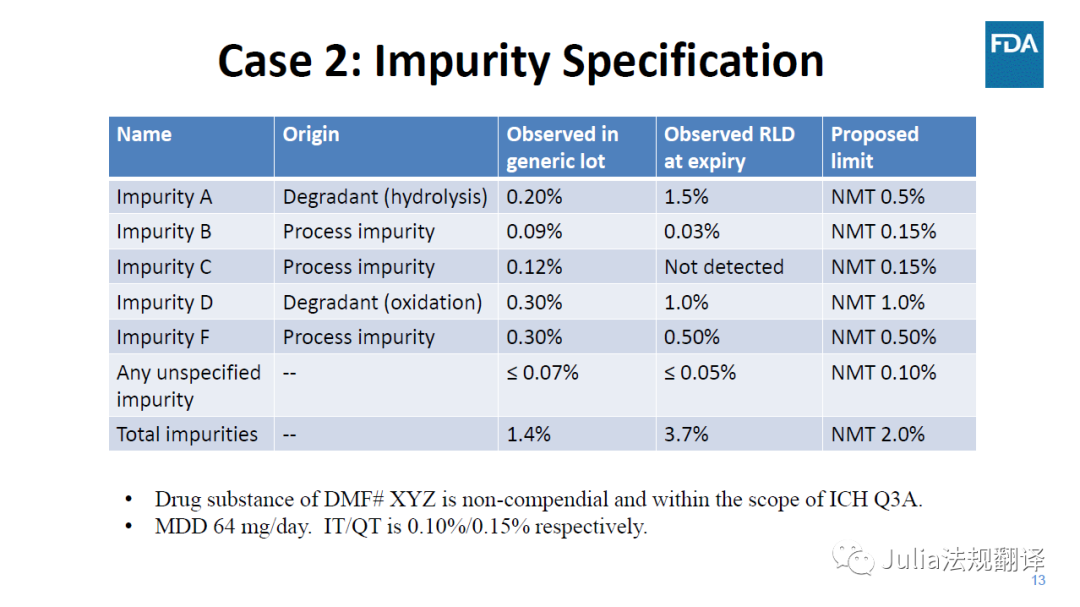
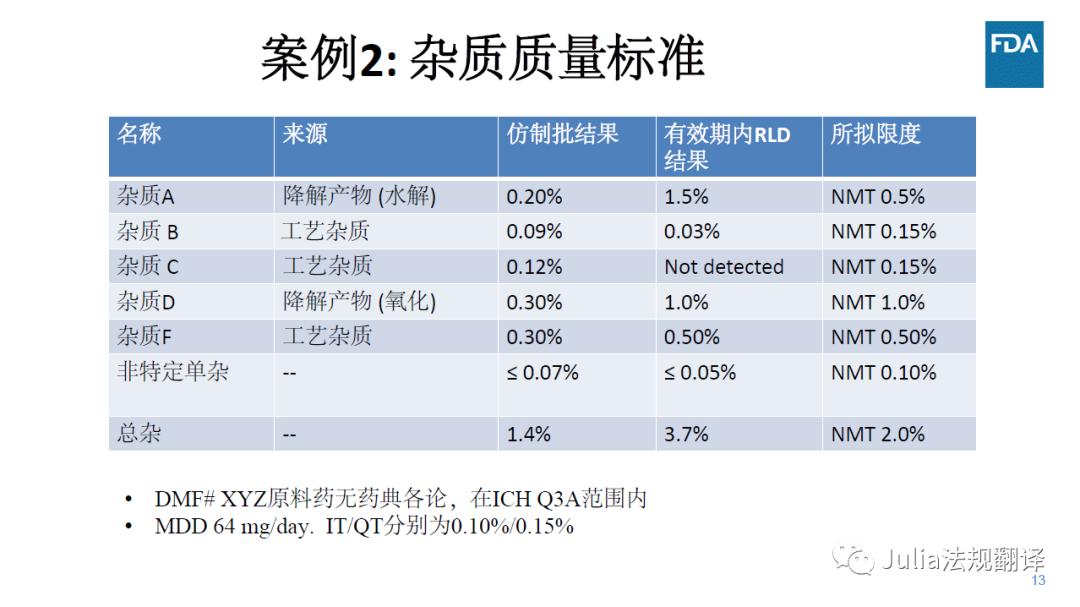
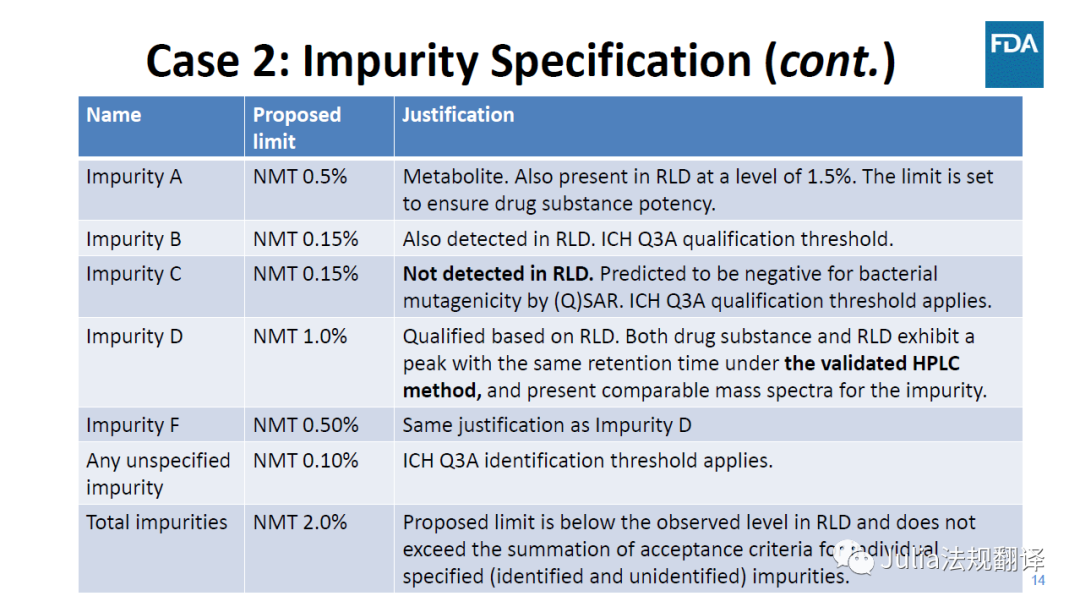
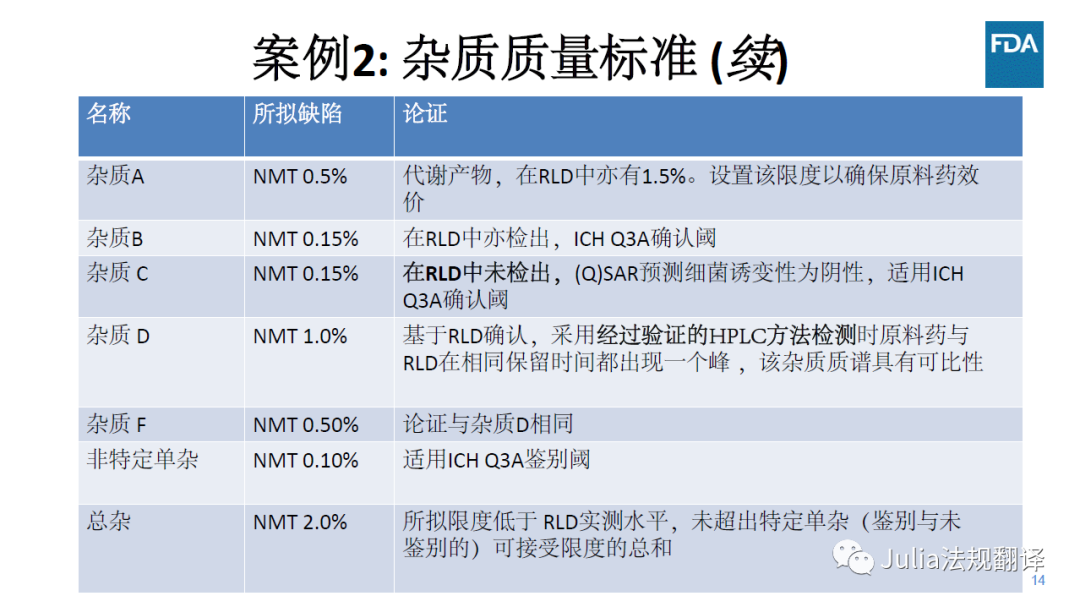
一 案例2:使用RLD论证杂质标准
-
残留溶剂
一 案例3:没有既定PDE的残留溶剂确认
-
元素杂质
一 案例4:矿物来源原料药
-
总结
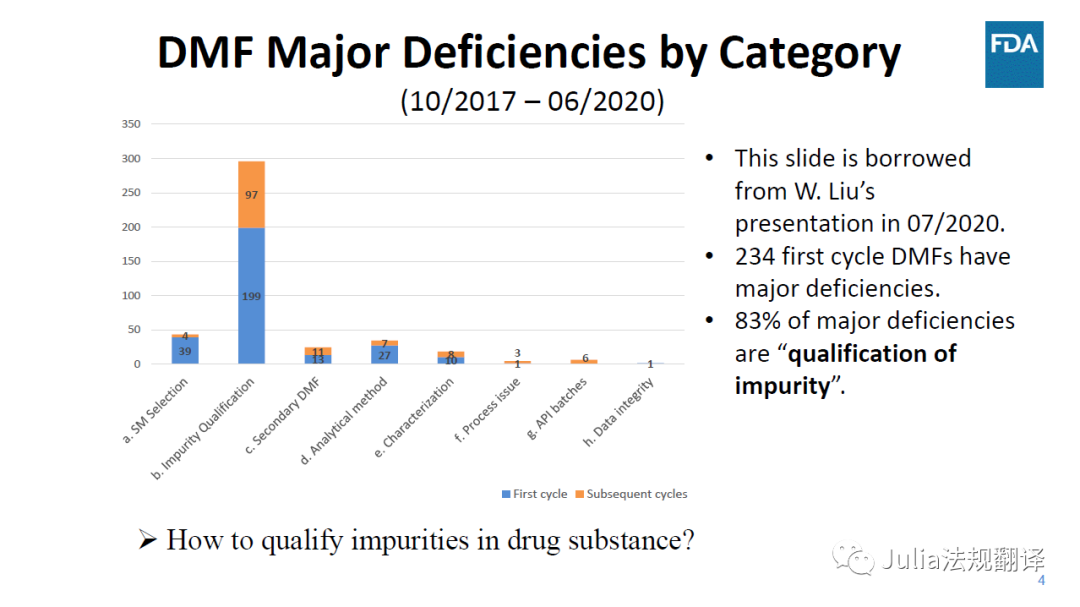
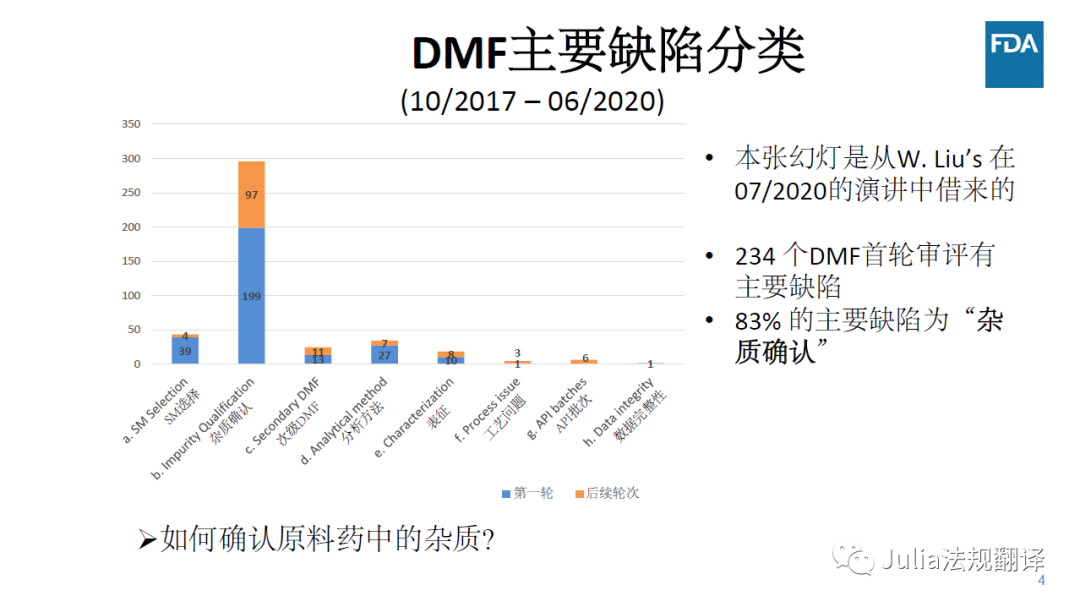

临床相关性
-
杂质可接受标准是在考虑到杂质水平的临床影响和危害评估的基础上建立的,这与依据生产工艺能力相反
-
对于不在ICH Q3A适用范围内的原料药,特定杂质的可接受标准
在ICH Q3A确认阈以下
一般是可接受的,前提是在该水平没有毒性、免疫或临床关注
-
可接受标准应来自于临床试验、非临床研究(例如,诱变性评估计算机模型、体外、以及动物研究)、使用条件、前验知识(例如RLD)、公开资料和分析能力(适当时)得到的数据
政策与程序手册MAPP5017.2《依据临床相关性建立杂质可接受水平作为NDA、ANDA和BLA质量标准的一部分》2020年5月

最大日剂量(MDD)
*行业指南《在ANDA申报资料中引用已批准药品》2010年10月




其它确认方法
-
杂质实测水平和拟定可接受标准不超过参比制剂中实测水平
-
一 分析方法应经过验证并具有稳定性指示性
-
一 直接与RLD是最佳方法。以下其它方法需要药学/毒性评估
-
杂质为原料药的重要代谢产物
-
杂质的实测水平与拟定接受标准有科学文献进行充分论证
-
杂质的实测水平与拟定接受标准
均未超过毒性研究中经过充分评估的水平


残留溶剂
-
ICH Q3C中有已知浓度限或既定PDE的溶剂
一 1类溶剂:应避免使用的溶剂
一 2类溶剂:应限制使用的溶剂
一 3类溶剂:低毒性溶剂,PDE
≥
50mg/天
-
没有足够毒性数据的溶剂
一 生产商应提供这些溶剂残留水平的论证
一 ICH Q3C提供了建立PDE的方法(附录3),计算要经过药学/毒性评估
-
在特定情形下可接受更高水平残留溶剂,例如短期(<30天)或局部用药
一 此类水平应各案论证

限度描述方法
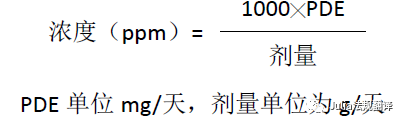

案例3:溶剂确认
-
二异丙醚(DIPE)类别为根据ICH Q3C没有足够毒性数据的溶剂
-
DIPE用于DMF#XYZ生产工艺较早步骤,实测水平为NMT5ppm
-
DMF持有人引用NIOSH(美国国家职业安全卫生研究所)资料拟定限度为1400ppm
-
MDD为4g,表明暴露值高达5600
μ
g/天
-
所拟限度是否影响审评时限?



元素杂质
-
ICH Q3D提供了一种评估和控制制剂中元素杂质的流程,该原则适用于原料药
-
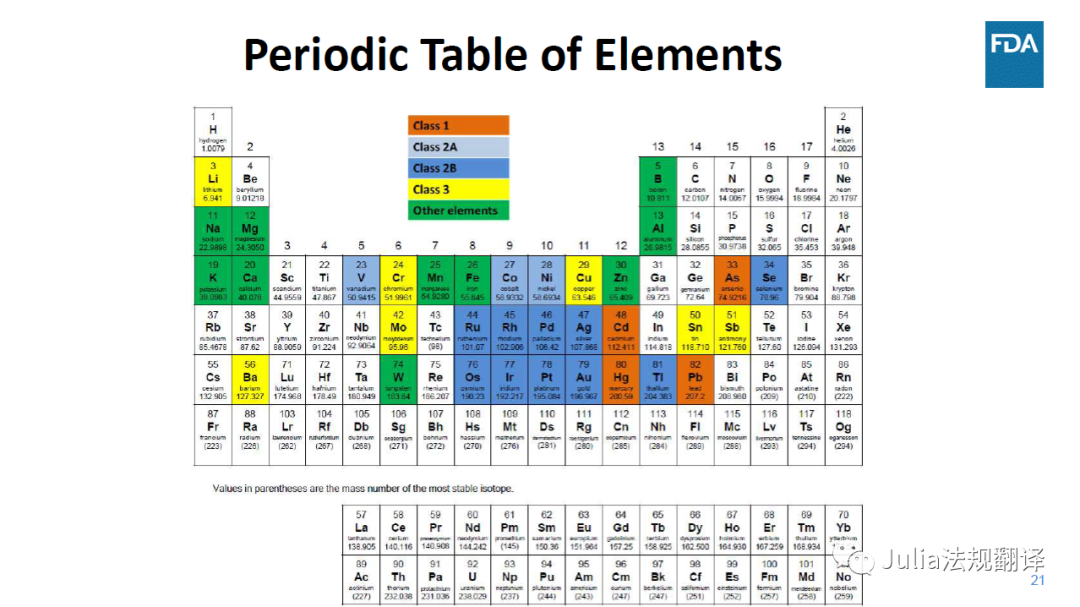
ICH Q3D中的元素杂质分类
一 1类:人类毒性物质,药品生产中受限或不能使用
一 2类:与给药途径有关的人类毒性物质。2A类元素发生可能性相对高,2B类元素发生可能性较低
一 3类:口服低毒性(PDE>500μg/天)
一 其它元素:低内在毒性但有其它指南和/或地方法规要求

风险评估
-
合成中有意加入的元素杂质(例如催化剂、试剂)必须受控或经过论证
-
支持多个MDD不同或给药途径不同的制剂申报的原料药按最严要求处理
-
强烈建立对原料药进行元素杂质风险评估
一 1类和2A类元素应评估
一 注射剂和特定PDE<500μg/天的吸入制剂应考虑3类元素

元素杂质的确认
-
1/2/3类元素,ICH Q3D中已确定PDE
一 假定MDD为10g或使用已知MDD计算浓度限度
一 使用其它给药途径者,可采用ICH Q3D附录1中说明的方法计算PDE,要进行药学/毒性评估
一 在特定情形(例如间断给药)下可以接受
高于既定PDE的水平
。PDE可按ICH Q3D附录1中所述方法各案计算,要进行药学/毒性评估
-
其它元素,ICH Q3D中未订立PDE值
一 低风险,使用特定质量考量者除外
-
ICH Q3D中未列出的元素
一 所拟限度不得超过RLD中实测水平
一 采用科学文献或毒性研究确认,要进行药学/毒性评估

案例4:矿物来源原料药
-
此类原料药通常采用矿物原料通过简单的水性系统生产步骤制得(溶出和沉淀或结晶)
-
来自自然污染的元素杂质和浓度可能会根据原料的来源而有很大变化
-
根据生产工艺知识进行风险分析可能不可行亦不可靠
-
元素杂质应当作“有关物质”处理,在原料药质量标准中进行常规控制










