太懒只看重点版:
1.
RNA-seq
技术重复很好,意味着测到即真实
?
A:
低丰度基因是测不准的,相应的,如果建库浓度过低,所有基因都测不准。
2.
转录组差异表达分析中应该如何对待超低丰度基因?
A:
过滤掉。
3.
超低丰度基因过滤标准是如何定的?
A:
似乎是拍脑门定的,你也可以用你自己的标准。
真粉必看长文版:
我们复杂细胞混合体,包括动植物,有一个很有趣的特点:虽然每个类型的细胞中
DNA
序列都几乎完全一样(体细胞中
DNA
突变率好像比我们想像的高)
,
但表达的
mRNA
却各有特色。其中的奥秘仍然等待着我们去探索。
正是
mRNA
的这种弹性,给了我们很多依赖
RNA-seq
造文章的机会,同时也带来了一个边缘性问题:有一些基因总是似有若无地表达,或者表达区域十分特异,在取样中被混合的其余细胞
mRNA
冲得很淡,比如只在生长点几个细胞内表达的干细胞基因,我们只能测到些痕迹。我们姑且统称这些基因为“
超低丰度基因”。
RNA-seq
结果中,
FPKM
从
0
到几万都存在,哪个部分算超低丰度基因?这个问题我不知道答案,因为任何答案都会显得很武断,很主观。
我们不如先迂回到另一个相关的问题:
1.
RNA-seq
技术重复很好,意味着测到即真实
?
RNA-seq
技术从诞生之初便是以技术重复性好著称的,技术重复性好的潜台词相当于“我测得每一个数据都是接近真实的”。早期曾经一度有公司拿着这个特点作招牌怂恿拮据的研究者不做生物学重复,催生了好几篇高水平期刊论文专门说明这个每个研究者其实心知肚明的小问题(技术重复再好也不能代替生物学重复,因为它们针对的是不同类型的误差)。
根据我自己的经验,我坚定地承认
RNA-seq
的技术重复性的确非常值得认可,但“我测得每一个数据都是接近真实的”是不是真实的呢?
我们可以用两个实验来验证这个猜想,(
1
)对相同的
mRNA
样本连续测两次,比较一下两次结果的相关性;(
2
)把
mRNA
稀释不同浓度,比较本技术对样本起始浓度的鲁棒性。
来看结果:

两次技术重复间相关性很高,基本上是一条线。但当
read count
越来越小时,这种相关性变得越来越差,数据越来越分散。
红色的线是我画上去的
,我个人觉得在
read conut >10
的时候,情况还是比较好的。
当起始
mRNA
减少后会如何呢?比如稀释到低于我们通常建库浓度时:
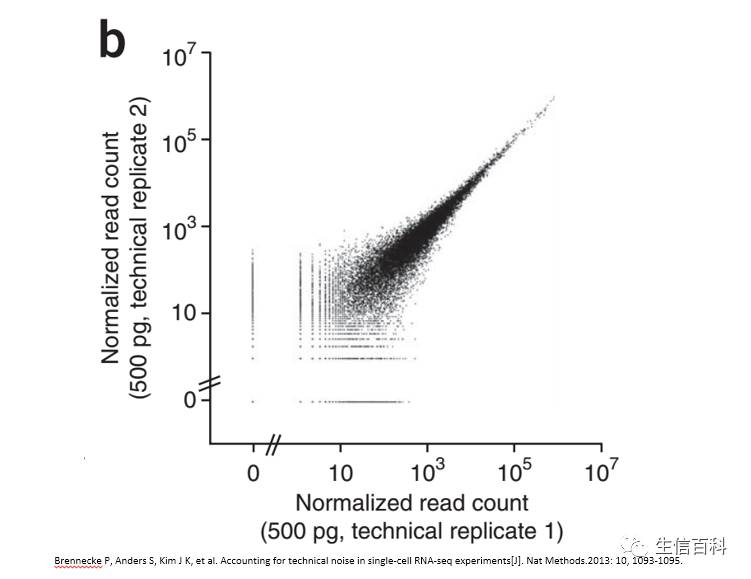
情况好像变得更严重了!这一部分被稀释的基因表达丰度变低了,丰度变低,离散度便同进增加。
再稀释一些如何?
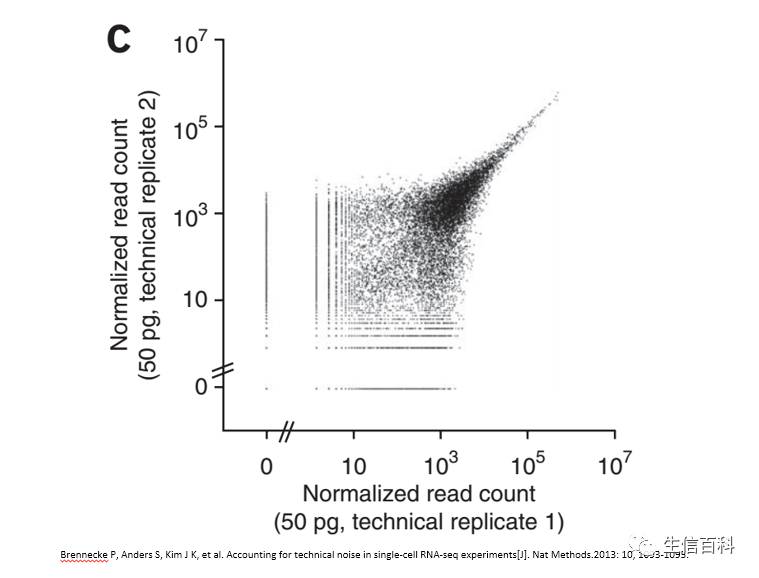
这就有点故意刁难
RNA-seq
了,这个浓度下,相当时大部分基因都是低丰度基因,情况已经不能接受了。
看来,
RNA-seq
的技术重复性好也是有条件的,低丰度基因测不准!
2.
转录组差异表达分析中应该如何对待超低丰度基因?
低丰度基因测不准对于差异表达基因筛选可是个大事,因为
1
与
0.001
之间
FC
是
1000
啊,这么诱人的数值,非常可能是假的?哪还了得!
那我们该怎么办呢?
实际上有部分软件要求过我们提供“过滤过的数据”,只是我们通常都不会在意,所以其它软件干脆默认进行过滤。
比如在运行
sleuth
时,它会告诉你:
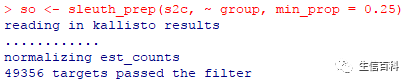
其中有一行:
“
49356 targets passed the
filter”,
也就是告诉你,我只对这些基因进行差异表达计算。
我去!你什么鬼参数干掉我
2
万多个基因?
大家看我的代码中有一个“
min_prop=0.25
”参数,这个参数在软件推荐参数中是没有提到的,扒出它的原代码,可以看到它的默认值是
0.47
。
这个参数的功能是“在所有样本中,只有
47%
的样本中本基因
map
到的
read counts >5
,这条基因才被计算”,比如你有两组样本,每组
3
个重复,至少有
3
个样本中本基因
read counts>5
,它才能通过。
为什么是
47%
?作者只是想保证那些只在一组样品里表达的基因仍然可以被筛选到,
48%
也可以,
49%
当然也可以。我把它改成了
25%
,因为我有
3
组样品。
为什么要定
read counts >5
?我估计作者也不知道,我个人觉得这个标准相当低。不过回到我们最初的起点:任何答案都会显得很武断,很主观。
测不准就过滤掉,够简单够粗暴,测到它们只能算运气好。
要想不错过重要基因,取样组织越纯粹越好。别嫉妒人家弄个单细胞
RNA-seq
可以上
Nature
,这很重要呢。