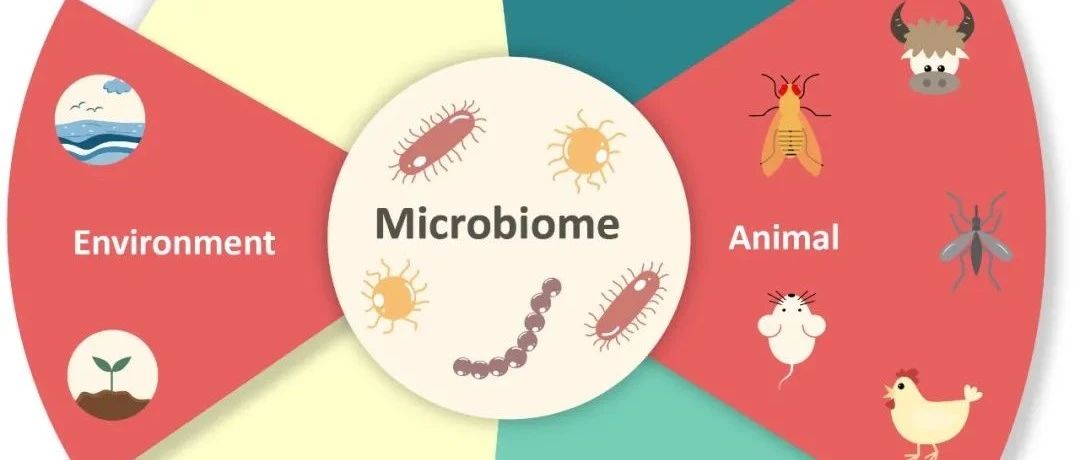
通过R-2HGxFTOxm6A轴抑制CEBPA抑制FTO转录
作者还发现延长R-2HG处理(例如96小时)可以显著降低敏感性白血病细胞中的FTO蛋白水平,但在耐药细胞中没有发生这种情况(图5A)。R-2HG处理48小时显示出对FTO蛋白水平的较小影响,但poly(A)+ RNA中的全m6A丰度明显增加(图5B),
这意味着延长的R-2HG处理后抑制FTO表达可能是m6A丰度增加的结果。延长R-2HG处理介导的FTO下调可能归因于转录抑制(图5C)。为了鉴定可能调节FTO转录的转录因子(TF),作者分析了FTO和92个TF基因之间在四种AML数据集中在FTO基因的“核心启动子”中具有推定结合位点的表达之间的相关性(图5D )。其中,CEBPA,MZF1,NFATC3和RFX3显示正相关(p <0.05),而NFKB2和TFEB在所有四个数据集中呈现负相关(p <0.05)(图5E)。在4个正相关的TF中,只有CEBPA显示出在敏感细胞中R-2HG处理后m6A修饰增加和表达降低(图5F和5G)。
与R-2HG介导的效应一致,CEBPA的表达也可以通过过表达FTO并通过FTO敲低来实现(图5H和5I)。通过YTHDF2可以识别R-2HG处理后CEBPA mRNA增加的m6A修饰,导致CEBPA转录物稳定性降低和表达降低(图5J和5K)。此外,CEBPA在AML患者中异常上调(图5L),其强制表达促进了FTO启动子的转录活性(图5M)。在R-2HG敏感的白血病细胞中,敲低CEBPA减少细胞生长,减少FTO丰度,并大部分消除R-2HG诱导的生长抑制效应(图5N-5P)。总之,R-2HGxFTOxm6A轴抑制CEBPA,作为一种反馈机制可以进一步抑制FTO转录(图5Q)。
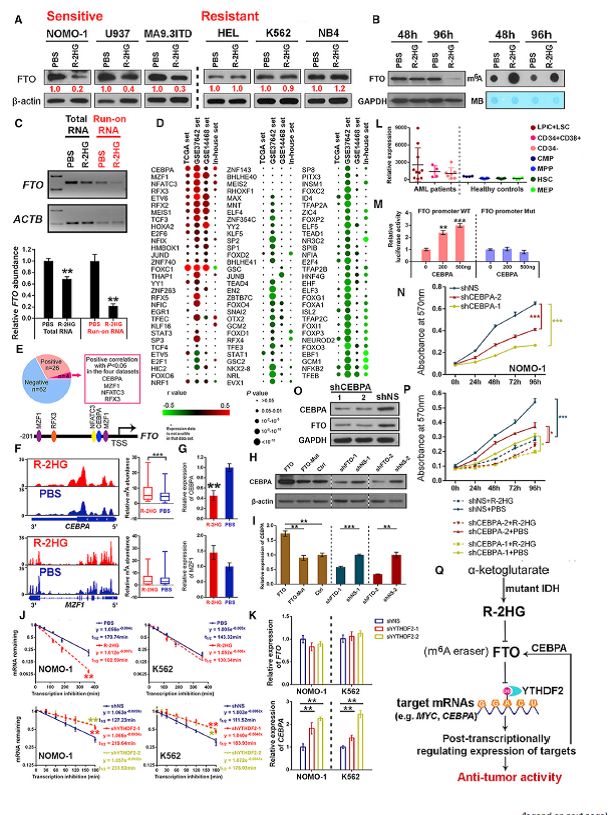
异位IDH1突变体和S-2HG都对R-2HG产生类似作用
为了确定突变体IDH是否可以模仿作者在R-2HG处理的细胞中观察到的表型,作者创建了具有诱导型突变IDH表达的白血病细胞系。诱导的IDH1R132H表达能模拟由外源R-2HG引起的表型,诸如抑制FTO和MYC表达,增强的m6A修饰,诱导的细胞周期停滞,减少的细胞增殖以及增加的细胞凋亡(图6A-6C)。细胞内R-2HG的测定证实IDH1R132H产生R-2HG至由300mM R-2HG处理产生的水平(图6B)。
S-2HG是R-2HG的对映体,它在结构和化学上与α-KG和R-2HG相关(图6D)。作者发现S-2HG在R-2HG敏感细胞中显示出生长抑制效应,但在R-2HG抵抗细胞中则没有(图6E)。 S-2HG也直接抑制FTO的去甲基化活性(图6F,6G)。 IDH突变和S-2HG一起可以在很大程度上模拟R-2HG的作用,包括直接抑制FTO,增加的全m6A修饰以及降低的白血病细胞增殖/活力。
FTO / MYC稳态控制R-2HG灵敏度
因为异位表达的IDH突变体(例如IDH1R132H)显示出抗肿瘤作用,为什么它们仍然存在于10%-20%的AML病例中?为了解决这个问题,作者使用图2A所示的RNA-seq数据对TCGA AML微阵列数据集进行了综合分析。作者鉴定了IDH突变型AML样品(相对于IDH-野生型AML样品,在整个集合或正常核型子集中)富集的5个核心信号传导途径,以及R-2HG抗性白血病细胞(相对到R-2HG敏感细胞)和R-2HG敏感性细胞(与健康对照相比)(图7A)。具有IDH突变的原发性AML细胞对JQ1(一种MYC信号传导抑制剂)也敏感,IC50低于1 mM,但IC50高于野生型IDH的AML细胞(图7B),可能是由于前者中MYC信号传导的过度激活。作者还证实,与IDH野生型AML样品相比,IDH突变AML样品具有更高的MYC及其关键靶标(例如CDK4和CDK6)表达水平和更低水平的FTO(但不是ALKBH5)表达(图7C)。Western blot显示R-2HG抗性白血病细胞系比敏感细胞系具有更高水平的MYC和更低水平的FTO; R-2HG处理导致敏感细胞中FTO和MYC表达显著降低,但对抗性细胞的影响最小(图7D)。
因此,作者认为高度活化的MYC信号传导可以降低R-2HG在大多数IDH突变AML细胞中的抗白血病作用。在抗性细胞中,诱导MYC敲低和JQ1介导的MYC抑制都可以增加它们对外源R-2HG和IDH1R132H的敏感性(图7E)。在敏感细胞中,MYC的过表达确实使得白血病细胞对R-2HG或IDH1R132H具有抗性并挽救R-2HG或IDH1R132H诱导的增殖抑制(图7F)。
R-2HG和化疗药物之间的协同作用也在体内得到验证。 R-2HG加柔红霉素或地西他滨的组合治疗在用R-2HG敏感的白血病细胞异种移植的白血病小鼠模型中显示出更好的治疗效果(图7G和图7E)。
总之,作者的数据表明高丰度的FTO赋予白血病细胞R-2HG敏感性,而MYC相关通路的超活化使白血病细胞(包括具有内源性IDH突变的细胞)对R-2HG具有抗性(图7H)。此外,R-2HG与一线化疗药物及其组合在临床上对治疗白血病具有显著的协同抗肿瘤作用。