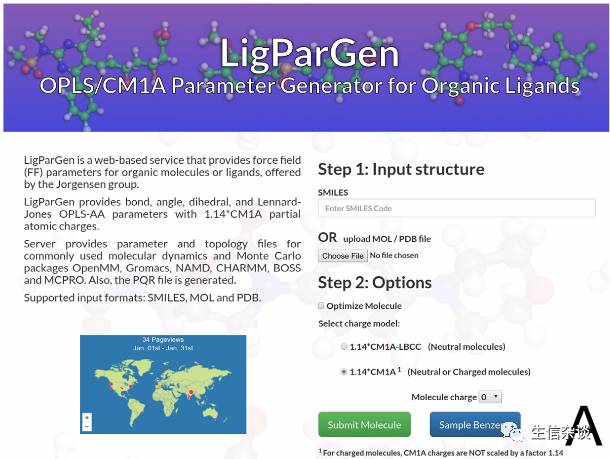
LigParGen是一个提供有机分子或者配体力场的在线服务网站,其能够提供键长,二面角以及Lennard-Jones OPLS-AA参数,电荷算法为1.14*CM1A或1.14*CM1A-LBCC。生成的配体力场可以非常容易的在Gromacs,NAMD以及OpenMM中使用,文章中说明将在不久提供Amber软件的支持。最大配体原子大小不超过200个原子,计算模型的电荷最大为正负2。
该在线服务器的使用方法非常的简单,并且使用的电荷计算模型1.14*CM1A在OPLS-AA力场中水化自由能计算(hydration free energies ,HFE)与实验具有良好的拟合效果,而改进的局部电荷校正(LBCC)方法更加提供了电荷的精确度,称之为1.14*CM1A-LBCC模型。在在线服务中,对于不带电的小分子提供1.14*CM1A和1.14*CM1A-LBCC模型,而带电荷的分子仅提供不放大1.14倍的CM1A模型。故带电荷的小分子还是最好使用GAFF力场制作方法。
LigParGen服务网址为:http://zarbi.chem.yale.edu/ligpargen/index.html
理论基础
CM1A方法使用来自于电荷密度的Mulliken分布分析,而电荷密度获得的方式为AM1方法基于配体几何计算,
故配体几何的差别会导致电荷计算轻微差别,但影响不大。
Mulliken电荷对原子A的计算方法大致如下(具体可以查看参考文献4):

q
A
是部分Mulliken 电荷,
Z
A
是原子A的中性电荷,
N
A
是原子A的电荷密度分布,计算方法如下:
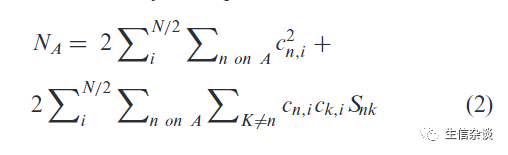
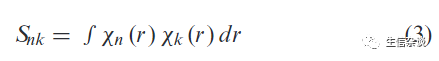
N
是分子的总电荷数,
C
n,i
为分子轨道系数,
χ
n
和
S
nk
QM多重积分,但1.14*CM1A电荷模型在苯环,醚类和酮类中参数不理想, 在426个有机分子的测试集中和实验数据相比mean unsigned error (MUE)达到1.5kcal/mol,而改进的局部电荷校正(LBCC)方法具有更好的效果。
作者进行了水化自由能(HFE)的评价,模拟的计算方法软件采用的NAMD,一共计算了10个分子,每个分子进行了36个窗口计算,每个窗口模拟1ns,力场采用OPLS-AA/1.14*CM1A-LBCC,水采用的TIP3P,完成参数请看底部的参考文献3。实验结果与模拟的结果拟合的较好,结果如下:
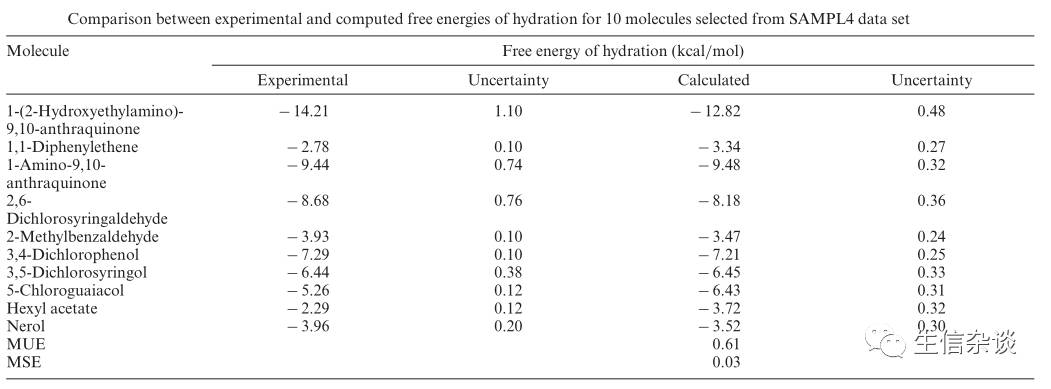
说明模拟结果和实验结果拟合的非常棒!
LigParGen使用方法
LigParGen的使用方法非常的简单:
首先上传MOL或者PDB文件或者SMILES Code:
当然也可以通过JSME自己画,如下图:
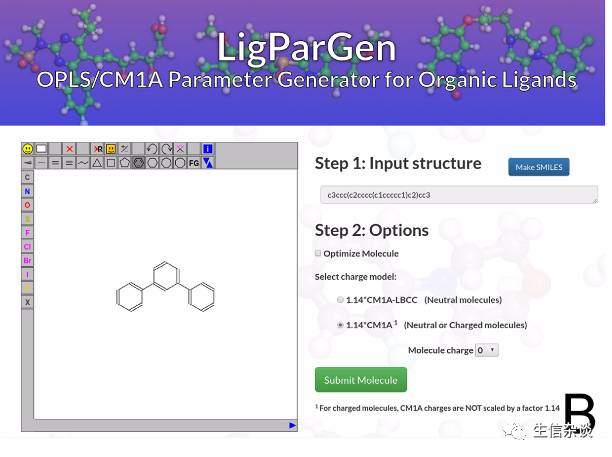
首先点一下Make SMILES,画完以后再点击一下Make SMILES即可。
然后选择电荷模型,是否优化分子,分子电荷等选项。
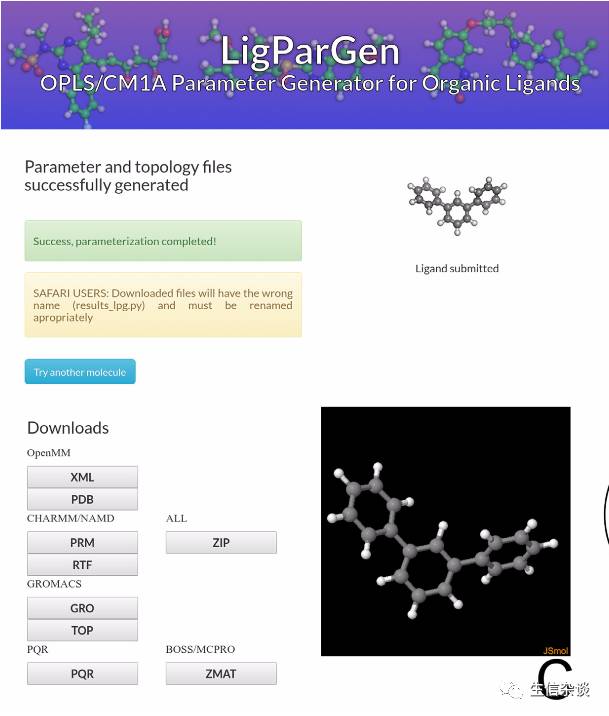
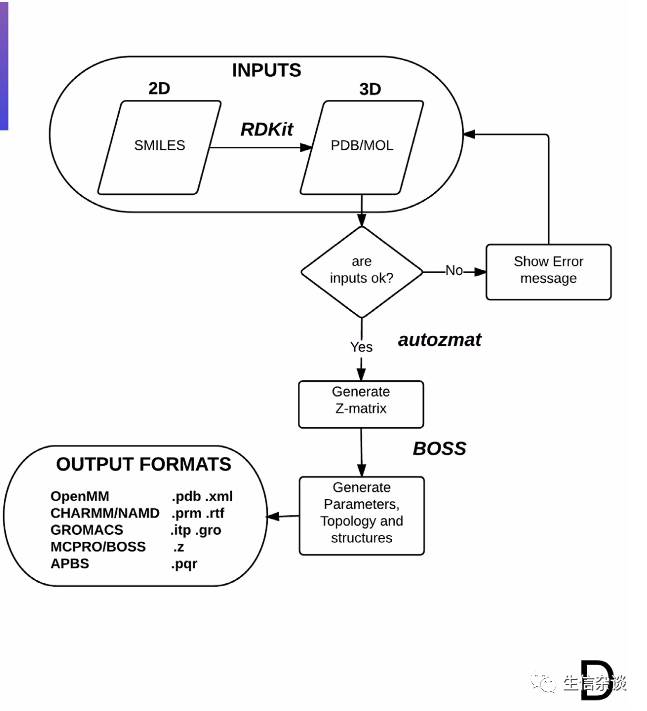
之后便可以下载不同软件需要用到的文件。软件的工作流程如下:
蛋白配体复合物模拟
LigParGen提供了3种模拟软件的蛋白配体复合物模拟的方法,可以在官网上查看,我们这里翻译GROMACS的蛋白配体复合物模拟。
1.准备蛋白配体系统
Gromacs,Chimera和python被要求安装。
1.1 获得复合物
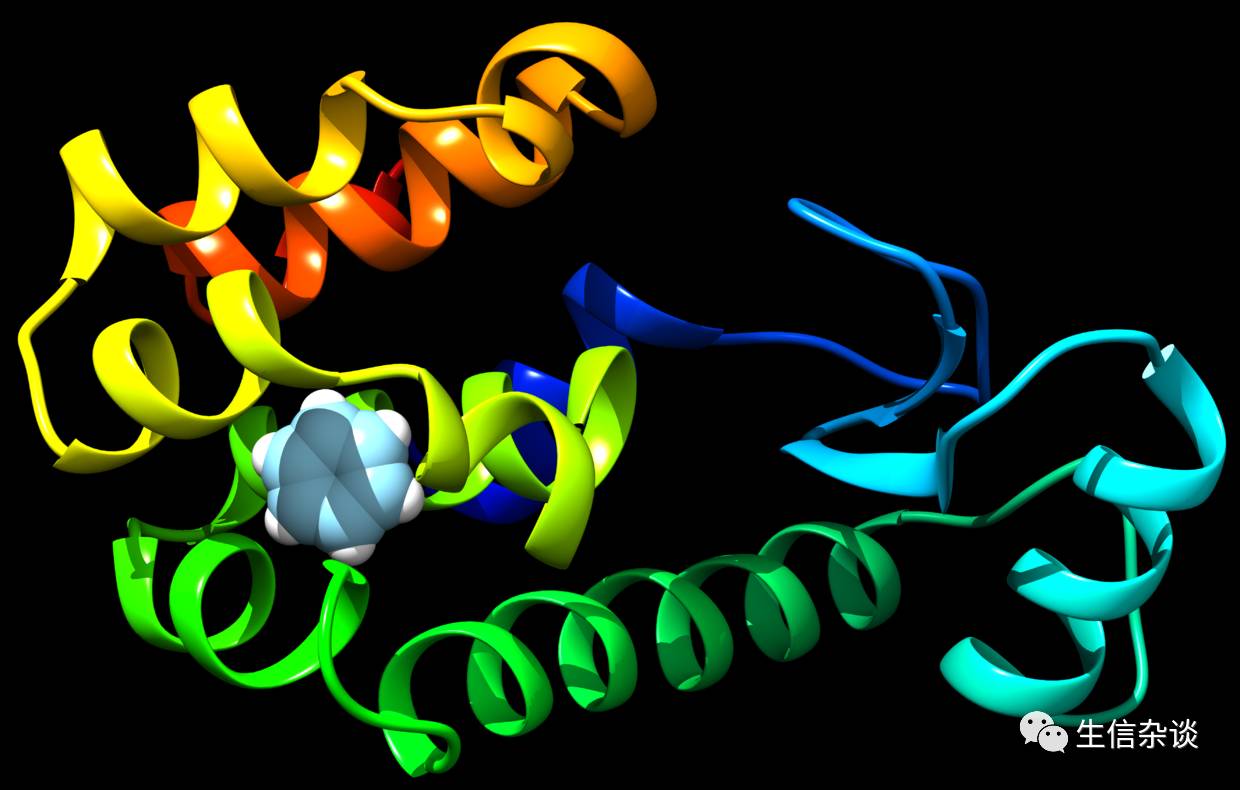
使用T4溶菌酶L99A与苯环分子复合物(PDB ID:4W52)作为教程,结构如下图,仅包含一条链
1.2 模拟前的蛋白准备工作
清理蛋白使用Chimera的对接准备功能(dockprep ),增加缺失残基使用Dunbrack rotamer库,这一部并不会进行蛋白加氢,因为在后面pdb2gmx会对蛋白进行加氢。Chimera脚本如下:
import chimera
from DockPrep
import prep
import Midas
import sys
import os
PDB_file = sys.argv[1]
lig_name = sys.argv[2]
os.system('grep ATOM %s > %s_clean.pdb'%(PDB_file,PDB_file[:-4]))
os.system('grep %s %s > %s.pdb'%(lig_name,PDB_file,lig_name))
protein=chimera.openModels.open('%s_clean.pdb'%PDB_file[:-4])
ligand=chimera.openModels.open('%s.pdb'%lig_name)
prep(protein,addHFunc=None,addCharges=False)
prep(ligand)
Midas.write(protein,None,"protein_clean.pdb")
Midas.write(ligand,None,"ligand_wH.pdb")
使用脚本:
Chimera --nogui --script "prep_prot_lig.py 4w52.pdb BNZ"
生成的 protein_clean.pdb和ligand_wH.pdb将会在随后使用。
1.3 准备配体
使用的方法如第2部分,后下载BNZ.gro(坐标文件)与BNZ.itp(拓扑文件)
2 设置Gromacs模拟
2.1 结合蛋白配体坐标
其Gromacs版本使用的为4.6.5
,若使用的为5.x或者2016版本可以具体查看李继存老师翻译的配体蛋白教程的后续命令。差别不大。
pdb2gmx -f protein_clean.pdb -o protein_processed.gro -water spce
两个蛋白的结合可以手动或者使用如下脚本完成:
import sys
pro_gro = sys.argv[1]
lig_gro = sys.argv[2]
pro = open(pro_gro,'r').readlines()
lig = open(lig_gro,'r').readlines()
pro_dat = [line.rstrip() for line in pro]
lig_dat = [line.rstrip() for line in lig]
tot_num = int(pro_dat[1])+int(lig_dat[1])
com_dat = [pro_dat[0],'%5d'%tot_num]+pro_dat[2:-1] + lig_dat[2:-1] + [pro_dat[-1]]for line in com_dat: print line
运行脚本
python combineGro_prot_lig.py protein_processed.gro BNZ.gro > complex.gro
2.2 蛋白配体拓扑结合
我们对拓扑文件(topol.top)进行修改。
在
#include "oplsaa.ff/forcefield.itp"
后加上
#include "BNZ.itp"
行来merge蛋白和配体。在
Protein_chain_A 1
后加上
BNZ 1
2.3 设置MD模拟
后面的部分与
载脂蛋白模拟
部分类似:
首先增加水盒子
editconf -f complex.gro -o complex_box.gro -c -d 1.0 -bt cubic
填充水:
genbox -cp complex_box.gro -cs spc216.gro -o complex_box_wSPCE.gro -p topol.top
增加原子:
grompp -f MDP/ions.mdp -c complex_box_wSPCE.gro -p topol.top -o ions.tpr
genion -s ions.tpr -o complex_box_wSPCE_ions.gro -p topol.top -pname NA -nname CL -nn 8
能量最小化:
grompp -f






