第1章 无处不在的生物信息学
说起生物信息学,好多未踏入此行的感觉好难,经常会问:有没有入门的资料呀,怎么学习生物信息学呀,我又没有计算机基础......
记着读11年读本科的时候,上了一门生物前沿课程,初步结识了生物信息学这一领域,但当时对其中的一些概念,一片迷糊。印象最深刻的就是那个Read的了。什么意思呀,读书?阅读?查了查词典,没其他意思呀,但就是搞不明白。无奈糊里糊涂度过了本科,以致于对那个时候正在火热的二代测序了解甚少。
虽然二代了解的少,但对于一代,小编我当年还是可以的。现在谈生物信息学,好多人直接就定义为高通量测序了,实际上生物信息学的根还是在一代Sanger测序分析上,所以学学习生物信息学,一代相关知识是必须要了解的。记着当年我大一下学期的时候进入了一个重点实验室学习做实验,算是我的生物信息学入门之处吧。在那里跟着学长学姐们学习提取DNA、RNA、质粒,然后PCR,跑胶等等的,学了好多实验东西(以致于小编在大四的时候写了一本新手入门的实验操作手册,不少内容小编前面已经发过,嘻嘻,参见链接:
生信人巨著|基础分子生物学实验操作指南
),同时也接触到了第一个生物信息学软件--引物设计软件primer premier 5.0(用法参见:
手把手教你设计PCR引物
)。实际上这个软件就涉及到了好多生物信息学知识,其中比较主要的就是比对的思想了。一条引物能够结合到模板的什么位置,主要就是通过比对实现的。哪个地方跟我这个引物最像,我这个引物就可能结合到这个地方。但同时我们又了解到我们设计的引物虽然跟这个地方很像,但又有一定的错配率和空缺率,这实际上就是比对算法中的错配(比对算法中常引入identity这个定义来衡量序列的相似性)和gap(gap意思就是匹配的区域模板多了个碱基,然后咱的引物序列在对应位置形成一个空位,称为gap;反之亦成立,只不过gap是位于模板上了)。说了这么多比对方面的东西,那个时候可能我根本没想这么多,一心可能只想通过反复的点点点,找个最佳的引物。
引物设计对于做实验的小伙伴们一般都非常熟悉。但其上游的东西,比如我的模板怎么来的,好多小伙伴可能就不熟了。因为获取模板序列这一部分是比较重要的,我们的老师们给咱提前弄出来了,我们实际过程中可能就不需要做此步了。但那个时候的我为了把我的的宏伟著作(哈哈)丰富了,我不断查阅文献,基本了解到了序列是怎么来的--NCBI。NCBI是啥东东,查了查资料,真有不少介绍(小编之前推送:
NCBI介绍
),其中我需要了解的就是从NCBI如何下载已知序列。查到了一篇别人写的方法(http://wenku.baidu.com/view/87d1dded551810a6f52486e2.html),比较详细,照着做终于会下序列了,那个时候感觉很兴奋。下载的序列后缀名是.fasta格式,对于没有接触生物信息学的我首先就查了查用什么软件打开这种格式的文件。软件很多,但实际上.fasta后缀名并不是我们普通认为的像.doc .xls,.mp4这样后缀名需要专业的不同的办公软件打开,他其实是一种文本文件,简单说就是.txt文件。包括还有的一些常用的生物信息学格式文件如fastq、gff3、sam,bed文件都是文本文件,可以用常用的办公软件打开,标注不同的后缀名主要是基于里面内容格式的不同,文件的文本属性没变。像fasta以>开头,fastq为四行一个单位,主要是二代测序原始数据的格式。
扯了这么多,序列下载完,然后接着要做多序列比对找同源区域设计引物。多序列比对见老师用clustalx(
基础工具-Clustalx用法
),看相关文献MUSCLE也挺好,于是我就下MEGA软件(里面内置了MUSCLE,具体教程看:
MEGA教程(一)
,
MEGA教程(二)
,
MEGA教程(三)
,
MEGA教程(四)
),准备比对。这里就涉及多序列比对和两序列比对了。多序列比对是找所有序列的最大交集,引申到生物学上就是找所有序列都保守的部分,这段序列在所有序列中可能是固定不变得,因而在无参考基因组条件下用此段序列当做模板设计引物效果会较好。两序列比对就是找两个序列最相似的部分,不能保证所有基因都有这段相似的部分。
设计好引物就开始P吧。P出来就要送去Sanger测序了。Sanger测序可谓黄金测序(
生信人16年浏览量最高文章推荐-黄金测序
),其测序读长也就一千来个碱基,所以基因长的就需要测两端然后拼接才能形成咱最终的想要的序列了。拼接的原理是什么,那就是overlap了(通俗说就是借助相同的序列把两端序列连在一起)。二代测序也就测个100多bp,相比一代短的要命,一个读长所读的序列就是前面所说的read了。为啥测得这么短,主要还是酶不给力。虽然短,但是二代测序普遍是高通量的(测个几十、几百个G bp很正常)就能够弥补短带来的缺点。当然现在三代读长十几个Kb,比一代还长,但引入错误也相对较对,需要增加数据量利用多序列比对来纠正错误,也还是可以的。关于测序原理,小编之前一篇浏览量极高的推送请参考:
测序简史
拼完序列就要注释一下,简单方法就是NCBI在线blast(
NCBI在线BLAST用法详解
)。blast原理要大体明白,其目的就是找相似的部分,判断我的序列与人家的已发表的序列相似性,具体原理生物信息学书都有介绍。
总之小编上面阐述的主要意思是生物信息入门较为简单的第一方向是把相关的window软件会操作,理解一些其中的参数及输出结果,然后再搞一些比较复杂的命令行软件及二代测序分析就容易了。
第2章 网络资源
了解了一些window软件,小编接着阐述一下生物信息领域中的网络资源。生物信息分析也可以称为大数据分析,而提大数据那就可以进一步延伸到数据库了,不断地数据积累做成了数据库,其中大家比较熟悉的就是NCBI了。那么了解这些数据库的用法,挖取对自己有用的数据那便成了关键。因此对于生物信息学初学者首先要认识的就是我要了解有哪些数据库,知道如何去用。
罗静初老师那次来讲课介绍了他们北大的一个网站abc bioinformatic(http://abc.cbi.pku.edu.cn/),上面有好多我们初学者需要了解的。对于生物信息学,对于我们初学者来说应该就像这网站名,要从abc一点一滴开始去学习。
废话不多说,小编在此列出几个重要的网站,希望大家能够去了解:其中工具集表示此网站有众多在线或者本地分析工具,数据集表示存储大量已发表的序列及其他生物信息学数据。
1、NCBI:工具集+数据集
NCBI介绍
2、EBI:工具集+数据集
网址:
http://www.ebi.ac.uk/services
3、
ExPASy:工具集
网址:
http://www.expasy.org/
4、Omics Tools:工具集
网址:
https://omictools.com/
5、CBS :工具集
网址:
http://www.cbs.dtu.dk/services/
6、CABRI:工具集
网址:
http://www.cabri.org/
7、SMA:工具集
网址:
http://www.bioinformatics.org/sms2/index.html
8、Ensemble:工具集+数据集
网址:
http://asia.ensembl.org/index.html
9、Softberry:工具集
网址:
http://linux1.softberry.com/berry.phtml
每个网站有各自的特点,资源含金量都相当高。对于我们初学者来说当我们遇到问题首先知道有这些资源我们可以去查,并且更重要的还要熟悉这些网站的用法。那么如何来熟悉网站用法?或者说如何使用在线工具(避开敲Linux代码)?这对于我们入门至关重要!
小编认为学会使用一个东西必须要动手,不能只看,而是带着问题去解决,去学习!比如说,一篇做基因家族分析的文章,首先他说从NCBI找到了300已知的MAPKKK基因家族成员,把他下载下来了。简单的一句话但这里面需要我们学习的很多,假设我们要模仿他做,那么我们就想这些已知的是如何在NCBI 中下到的,我们就去查,查到可以在nucleotide数据库中下到我们就输入MAPKKK结果出来了一大堆,联想是做植物的我们就只过滤植物的,加一步过滤选项,结果就OK 了,在这个过程中我们学会了如何使用NCBI 的nucleotide数据库,而不是漫无目的的看这个数据库的用法,总之一点我们要按需来学习,多去实战,在实战中学习,我们会进步很快。
再有就是我们要做结构域预测,文献中说是这两个网站(Pfam:http://pfam.xfam.org/和CDD:https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi),那么我们就挑个序列上去戳弄戳弄,用不了多少时间我们就搞明白了用法,理解了结果,这样一来我们后面批量操作本地版本的结构域预测软件就容易了,知道如何输入输出,如何设置参数。
最后就是好多初学者不知道用什么工具分析,或者就是相同的工具有好多个不知道选用哪个,这个就要根据你的研究目的来选,因为每个工具都有他侧重的一面,建议大家看最新发的工具,往往他会有跟前面工具的对比,顺便提到各工具的适用性。
第3章 生物信息学入门书籍
小编主要介绍一些入门书籍,并有电子版可供下载(
链接: https://pan.baidu.com/s/1hrC2LKS 密码: piyg
)!
1. 基因组与转录组相关的书籍
这本书比较全面系统的介绍了 DNA、蛋白质序列和结构、基因组、蛋白质组、转录组和系统生物学内容,也分别对原核生物、真核生物、人类基因组结构和特性进行了介绍和比较,并将基因组变化和进化联系起来。
此书详细介绍了转录组测序的相关技术,以及转录组测序后结果分析的详细的protocol,是做转录组分析必看的一本书!
2. 生物信息分析相关工具
生物信息分析大多数软件都是在Liunux系统下运行,因此必须学会Linux操作系统。《鸟哥的Linux私房菜》第3版是最具知名度的Linux入门书,该书全面而详细地介绍了Linux操作系统,生物信息入门优先可以看第三部分,该部分主要介绍 Shell Script以及文字编辑器Vim,当然如果想进阶的话可以学习其它几部分。
小编之前关于Linux一些介绍:
生信公司内部资料linux大全(一)
生信公司内部资料linux大全(二)
生信公司内部资料linux大全(三)
3. 计算机语言
目前比较盛行python,但perl仍然也经常用,一些生物信息学软件也都是用perl写得。小编因此推荐以下两本:
此书翻译过来是笨方法学python,不用说绝对是入门好教材。
Learn Python The Hard Way
《Perl 语言入门》第六版,也就是大家所称道的“小骆驼书”。学完这本书基本可以满足生物信息的分析的需求,如果还需要进阶,推荐阅读俗称“大骆驼书“的《Perl高级教程》。
小编之前介绍的一些用法如下:
Perl基础教程--精华篇(一)
Perl基础教程--精华篇(二)
一步一步教你学习Perl(一)
一步一步教你写perl(二)
生物信息常常需要大量的数学统计分析,且大部分结果需要已图形的方式展示,这就需要有一定的统计能力和画图能力,目前用的最广且开源的语言就是R语言。
《R语言编程艺术》,该书从最基本的数据类型和数据结构开始,到递归和匿名函数等高级主题,由浅入深,讲解细腻,读者完全不需要统计学的知识,甚至不需要编程基础。
小编之前关于R的介绍:
R语言绘图——图形标题、坐标轴设置
R语言作图的小方法--作图布局与坐标轴的控制
如何利用R包qqman画曼哈顿图?
R语言与热图绘制等
4. 各种生物数据分析软件
《常用生物数据库分析软件》作为生物信息学经典图书,本书罗列了一些常用的生物数据分析软件,从安装到运行都有具体说明,非常适合入门级使用。
总之,上面的书籍都是入门的工具书,可以边用边学,会有助于你迅速入门。同时大家也应该积极阅读一些文献,尤其是一些protocol,这样能够及时掌握目前流行的分析内容及好的工具。
第4章 视频资源
小编收集了一些网络视频资源,大伙可以关注一下,尤其是对于周围没有熟悉分析的人员,自己孤军奋战的青年们,看一下视频,对于快速入门还是至关重要的。下面列出了两组,网络上肯定会有更多,大伙可以自己检索,或者有知道还有好的资源的,请留言共享奥。
1. 这组视频来源于生信菜鸟团,
网址如下:
http://i.youku.com/trainee
-
5行R代码搞定表达芯片数据处理-jmzeng
-
5行R代码搞定表达芯片数据处理-jmzeng-补充视频-必须要看
-
第二讲_生物信息学习资源介绍01_北大生物信息学公开课
-
第二讲_生物信息学习资源介绍02_宾夕法尼亚州立大学公开课
-
第二讲_生物信息学习资源介绍03_斯坦福大学-计算生物学
-
第二讲_生物信息学习资源介绍04_斯坦福大学-遗传生物信息
-
第二讲_生物信息学习资源介绍05_德国柏林自由大学研究生生物...
-
第二讲_生物信息学习资源介绍05_德国柏林自由大学研究生生物信息学课程
-
第二讲_生物信息学习资源介绍06_美国明尼苏达大学生信课件
-
第二讲_生物信息学习资源介绍07_TCGA的历年会议ppt共享
-
第二讲_生物信息学习资源介绍08_生物信息学网上资料持续收集
-
第二讲_生物信息学习资源介绍11_安德森癌症中心生物信息课程共享
-
第六讲_综合项目08_写一个做富集分析的网页
-
第三讲_百款常用生物信息学分析工具005_网页工具_WeGO进行GO...
-
第三讲_百款常用生物信息学分析工具005_网页工具_WeGO进行GO富集分析
-
第三讲_百款常用生物信息学分析工具032_基础工具_IGV
-
第三讲_百款常用生物信息学分析工具038_外显子相关_Bowtie2
-
第三讲_百款常用生物信息学分析工具053_转录组相关_tophat2
-
第三讲_百款常用生物信息学分析工具055_转录组相关_Hisat
-
第三讲_百款常用生物信息学分析工具064_转录组相关_RNA-SeQC
-
第三讲_百款常用生物信息学分析工具069_R语言软件_GOstats做p...
-
第三讲_百款常用生物信息学分析工具069_R语言软件_GOstats做pathway的富集分析
-
第三讲_百款常用生物信息学分析工具094_其它软件_根据表达数...
-
第三讲_百款常用生物信息学分析工具094_其它软件_根据表达数据对癌症进行分类
-
第三讲_百款常用生物信息学分析工具095_其它软件_安装matlab...
-
第三讲_百款常用生物信息学分析工具095_其它软件_安装matlab运行环境
-
第三讲_百款常用生物信息学分析工具096_其它软件_GISTIC2.0识...
-
第三讲_百款常用生物信息学分析工具096_其它软件_GISTIC2.0识别拷贝数变异区域
-
第三讲_百款常用生物信息学分析工具097_其它软件_R里面的生物...
-
第三讲_百款常用生物信息学分析工具097_其它软件_R里面的生物数据注释包
-
第三讲_百款常用生物信息学分析工具098_使用R包cgdsr来下载TC...
-
第三讲_百款常用生物信息学分析工具098_使用R包cgdsr来下载TCGA的数据
-
第三讲_百款常用生物信息学分析工具099_用GSEA来做基因集富集...
-
第三讲_百款常用生物信息学分析工具099_用GSEA来做基因集富集分析
-
第四讲_数据处理脚本实例01_perl_snp格式化
-
第四讲_数据处理脚本实例02_perl_覆盖度计算
-
第四讲_数据处理脚本实例03_perl_平均测序深度
-
第四讲_数据处理脚本实例04_perl_简并碱基
-
第四讲_数据处理脚本实例05_perl_允许错配
-
第四讲_数据处理脚本实例06_perl_多重循环
-
第四讲_数据处理脚本实例07_perl_点突变分类
-
第四讲_数据处理脚本实例08_perl_二分法查找
-
第四讲_数据处理脚本实例09_perl_爬虫
-
第四讲_数据处理脚本实例10_perl_mutation上下文
-
第四讲_数据处理脚本实例11_perl_模块
-
第四讲_数据处理脚本实例15_R_进度条
-
第四讲_数据处理脚本实例17_R_T检验
-
第四讲_数据处理脚本实例18_R_聚类
-
第四讲_数据处理脚本实例19_R_apply系列函数
-
第四讲_数据处理脚本实例20_R_批量下载
-
第四讲_数据处理脚本实例22_R_3D条形图
-
第四讲_数据处理脚本实例24_R_数据整形
-
第四讲_数据处理脚本实例25_R_dplyr包
-
第四讲_数据处理脚本实例27_R_shiny
-
第四讲_数据处理脚本实例-补-_perl_写富集分析
-
第四讲_数据处理脚本实例-补-_R_写富集分析
-
第一讲_生物信息数据处理环境搭建01_服务器基础知识
-
第一讲_生物信息数据处理环境搭建02_用虚拟机体验root权限
-
第一讲_生物信息数据处理环境搭建03_系统环境变量设置
-
第一讲_生物信息数据处理环境搭建04_软件安装简介
-
第一讲_生物信息数据处理环境搭建05_非root用户自己安装perl
-
第一讲_生物信息数据处理环境搭建06_版本安装并更新R
-
第一讲_生物信息数据处理环境搭建07_非root用户自己安装python
-
第一讲_生物信息数据处理环境搭建08_非root用户自己安装java
-
第一讲_生物信息数据处理环境搭建09_如何使用mysql数据库
-
第一讲_生物信息数据处理环境搭建10_用root用户来安装Lamp环境
-
第一讲_生物信息数据处理环境搭建11_各种脚本语言模块处理基础
-
第一讲_生物信息数据处理环境搭建12_使用编辑器加快编程效率
-
根据高通量测序数据来判断样本性别
-
画基因的外显子覆盖度图
-
看看Y染色体上面的基因在测序数据里的覆盖度和测序深度
-
人的hg19版本基因组的所有相关数据下载
-
如何下载TCGA计划的相关数据
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture1
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture2
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture3_1
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture3_2
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture4
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture5
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture6and8
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture7
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture9
-
生信菜鸟团-5行R代码搞定表达芯片数据处理-lecture10
-
生信技能树论坛帖子编辑高级技巧系列教程1
-
使用picnic对拷贝数变异检测芯片数据进行分析
-
使用进度条和并行计算来模拟计算赌博输赢概率
-
下载最新版GO,并且解析好
-
下载最新版的KEGG信息-并且解析好
-
用excel表格做差异分析-看差异原理
-
用firehose_get 来下载所有TCGA寄存在broad的数据
-
用limma包对芯片数据做差异分析
-
用Mutation-Assessor软件来看突变位点对基因或者蛋白功能的影响
-
用重抽样和主成分方法来做富集分析
-
主成分分析略讲
-
自动化出网页报告的R语言包-Nozzle
2. 优酷上的
http://list.youku.com/albumlist/show?id=23674335&ascending=1&page=1
-
(第二讲上)NCBI全球基因芯片共享数据库使用方法
-
(第二讲下)Affymetrix表达谱芯片数据的基础处理
-
(第三讲上)NCBI疾病关联遗传位点注释查询
-
(第三讲下)Affymetrix_SNP与CNV基础分析系统
-
(第四讲上)NCBI深度利用_Gene功能注释数据库的查询实践
-
(第四讲下)可变剪接在疾病关联基因研究中的新视角
-
(天津第一讲下)基因芯片信息分析基础方法
-
(天津第一讲上)基因芯片信息分析基础方法
-
(第五讲上)Affymetrix_HTA
-
(第五讲下)从DNA到RNA_从编码基因到非编码基因_生物信息解决之道
-
非编码RNA调控方式理论_相关产品
-
超高分辨率全基因表达分析系统
-
超高密度全基因表达分析系统_产品
-
遗传多样性和基因表达调控
-
非编码RNA调控方式_案例_2011年hepatology
-
生物信息学在疾病研究中的应用
-
肿瘤研究基本思路
-
基因功能和信号通路分析最新进展
第5章 软件安装
其实妨碍大家进行生物信息学学习的一个比较关键的地方就是编程了。生物信息学是大数据科学,这就要求必须具备一定的编程思想,会采用计算机程序从庞大数据中挖掘有效信息。这就要求我们会基础编程,并且更重要的需要我们精通的是会安装和使用生物信息学软件。
首先计算机编程这一块有时候也是比较重要,毕竟不能手工进行处理庞大的数据吧。其实编程这一块主要是为了结果过滤,毕竟软件出来的往往并不能满足自己想要的结果,这就需要对软件出来数据进一步深挖过滤,拿到真正对自己有用的数据。编程这一块有人推荐学perl,有人推荐Python,无所谓了,关键看你周围的人用什么编程,方便在遇到问题时能够及时的解决。如果你对perl感兴趣,我们前天发过Linux与perl的推送,相信这将是非常有用的资料,快速掌握Linux与perl。
编程虽重要,但小编认为对于初学者软件使用更重要。大家都是生物狗,软件一些参数用法结合一下生物学意义相对来说容易理解,但是可能对大家比较困难的是软件用之前的工作--软件安装。由于不同的软件需要的依赖(包括种类和版本)不同或者使用的是公用计算机集群你根本无权限安装,导致软件安装不成功。稀奇古怪的报错信息,对于生物狗们真好似一头雾水!好不容易有个好软件但是不能用!所以大家需要掌握一些软件安装的技巧与方法。
本处主要讲你没有权限安装方法,即安装到自己目录下面方法(有权限安装通用)
1、perl模块安装
我们运行一些Perl程序时经常出现找不到某个module。对于这种报错,缺哪一个就下载哪一个或者看看软件包有没有此模块直接给路径添加即可。
首先下载所需要的模块,像本处为Keith module,这样我们谷歌或者CPAN((http://search.cpan.org/))上下载Keith.pm即可。
运行命令:
perl Bin/trf_wrapper.pl
报错信息:
Can't locate Keith.pm in
@INC
(you may need to install the
Keith module
) (@INC contains:
/share/nas2/genome/biosoft/perl/current/lib//5.20.0/x86_64-linux-thread-multi /share/nas2/genome/biosoft/perl/current/lib//5.20.0
/share/nas2/genome/biosoft/perl/current/lib/)
at Bin/trf_wrapper.pl line 13.
BEGIN failed--compilation aborted at Bin/trf_wrapper.pl line 13.
解决方案:
只需要在
trf_wrapper.pl中调用的Keith模块(
use Keith;
)之前加入下面红色部分即可,其中PATH为模块
Keith.pm
所在的目录。
BEGIN{
push (@INC,"PATH/");
}
use Keith;
2、R包安装
我们运行一些R语言程序时经常出现找不到某个
package
。
对于这种报错,缺哪一个就下载哪一个。
首先下载所需要的
package
,像本处为
ggplot2
,这样我们谷歌或者
bioconductor
(http://www.bioconductor.org/)或者CRAN(https://cran.r-project.org/)上下载ggplot2即可。
运行命令:
Rscript heatmapV2.R
报错信息:
Error in library(ggplot2) : there is no package called 'ggplot2'
解决方案:
下载到ggplot2_2.2.1.tar.gz,然后用下面命令(针对无管理员权限,安装自己目录下)安装即可。
R CMD INSTAL
L
ggplot2_2.2.1.tar.gz
注意安装log:
installing to /home/xxx/R/x86_64-unknown-linux-gnu-library/3.1/
ggplot2_2.2.1
/libs
安装完成后添加环境变量:
export LD_LIBRARY_PATH=
/home/xxx/R/x86_64-unknown-linux-gnu-library/3.1/
ggplot2_2.2.1
/libs
:$LD_LIBRARY_PATH
3、Python包安装
注意使用的python版本,Python2与Python3差别较大,因此安装时注意python版本。
运行命令:
/Python/3.5.2/bin/python suppa.py
报错信息:
Traceback (most recent call last):
File "suppa.py", line 9, in
import fileMerger as joinFiles
File "/share/nas1/SUPPA-master/fileMerger.py", line 11, in
import pandas as pd
ImportError: No module named 'pandas'
解决方案:
找到对应版本的pandas下载即可,文件格式一般为后缀名为.tar.gz:
pandas-0.20.1.tar.gz
安装步骤:
tar zxvf
pandas-0.20.1.tar.gz
cd
pandas-0.20.1
python setup.py install --user
安装完成后注意在.bashrc中添加环境变量,
PYTHON_PATH=/home/xxx/.local/lib/python3.5/site-packages/:$PYTHON_PATH
4、C包
无root权限的linux系统上安装软件时候遇到的lib××× not found的问题.
参见此博文:http://blog.shenwei.me/solve-lib-not-found-in-linux/
第6章 实战
实战。顾名思义,就是要实际动手操作。但是对于初学者来说,可能手忙脚乱,不知道怎么去做,但是只要不断去尝试与探索,一切都会明了。
小编下面列了之前发的基因家族分析套路,可以作为初学者入门实战的非常好的材料,同时小编末尾附了几篇关于基因家族鉴定分析的中英文文献(大伙也可以自己去找基因家族鉴定的文献),里面描述相对详细,大家可以照着去做,看能否模拟出文章中结果,相信通过此过程,你必将会对生信有很好的理解与把握了。
一、基本分析内容
*数据库检索与成员鉴定
*进化树构建
*保守domain和motif分析.
*基因结构分析.
*转录组或荧光定量表达分析.
二、数据库检索与成员鉴定
1、数据库检索
1)首先了解数据库用法,学会下载你要分析物种的基因组相关数据(参见
如何下载一个物种基因组和GFF文件
)。一般也就是下面这些数据库了
Brachypodiumdb:http://www.brachypodium.org/
TAIR:http://www.arabidopsis.org/
Rice Genome Annotation Project :http://rice.plantbiology.msu.edu/.
Phytozome:http://www.phytozome.net/
Ensemble:http://ensembl.gramene.org/genome_browser/index.html
NCBI基因组数据库:http://www.ncbi.nlm.nih.gov/assembly/?term=
2)已鉴定的家族成员获取。
如何获得其他物种已发表某个基因家族的所有成员呢,最简单的就是下载该物种蛋白序列文件(可以从上述数据库中下载),然后按照文章中的ID,找到对应成员。对于没有全基因组鉴定的,可以下列数据库中找:
a. NCBI: nucleotide and protein db.
b. EBI: http://www.ebi.ac.uk/.
c. UniProtKB:http://www.uniprot.org/uniprot/
2、比对工具。一般使用blast和hmmer,具体使用命令如下:
*Local BLAST
formatdb–i db.fas–p F/T;
blastall–p blastp(orelse) –i known.fas–d db.fas–m 8 –b 2(or else) –e 1e-5 –o alignresult.txt.
-b:output two different members in subject sequences (db).
*Hmmer (hidden Markov Model) search. Thesame as PSI-BLAST in function. It has a higher sensitivity, but the speed islower.
Command:
hmmbuild--informatafaknown.hmmalignknown.fa;
hmmsearchknown.hmmdb.fas>align.out.
3、过滤。
*Identity: 至少50%.
*Cover region: 也要超过50%或者蛋白结构域的长度.
*domain: 必须要有完整的该蛋白家族的。工具pfamdb (http://pfam.sanger.ac.uk/) 和*NCBI Batch CD- search. (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi).
*EST 支持
*Blast and Hmmer同时检测到
4、通过上述操作获得某家族的所有成员
三、构建进化树的基本步骤
1. 多序列比对. Muscle program.
2. Model 选择. 分别针对蛋白序列和核酸序列的模型选择程序。ProtTest program for protein and ModelTest or Jmodetlest for DNA(http://user.qzone.qq.com/58001704/blog).
3. 算法选择。三种. NJ, ML and BI。
一般ML树比较准确,但应结合方法,如NJ树,相互验证。
4.软件选择。
MEGA (bootstrap least 1000 replicates),
phyML and Mrbayes (http://user.qzone.qq.com/58001704/main).
5.进化树修饰.
MEGA:view->options and subtree-> draw options. Also can be decorated in word
(http://user.qzone.qq.com/58001704/main)
具体步骤
1.多序列比对。一般采用muscle。因为 MUSCLE is one of the best-performing multiple alignment programs according to published benchmark tests, with accuracy and speed that are consistently better than CLUSTALW.
2 模型选择。
对于用蛋白序列构建进化树的可以采用下面命令:
java -Xmx250m -classpath path/ProtTest.jar prottest.ProtTest -i alignmfile.phy.
运行结果如下图
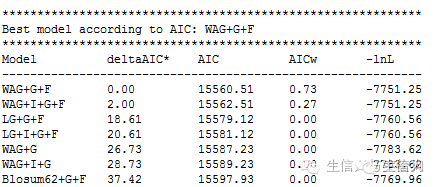
注意:
1)“.Phy” format. Only allow ten charaters.注意名字不能重复相同。









