每年 DIA 盛会,最让人关注的就是 CFDA 专场,场下座无虚席,连楼道中都已人满为患,而讲解的内容则完全可以用字字珠玑来形容。
结合现场参会讲者先后出场顺序,我们一起来做个简要回顾。
审评审批制度改革——药品
注册司李金菊副司长
李司长会上提到了药品审评审批制度改革主要目的是:
1、提高药品质量
2、提高效率
3、鼓励创新
4、公开透明
李司长同时就各项法规的进展做了相关介绍。

药品
注册司李金菊副司长
李司长提到,截止 2017 年 3 月,按照新分类受理化药注册申请共 542 件,其中创新药 184 件,占总量的 42.8%,中药和生物药的注册分类改革征求意见有可能也会于近期出台,事实上在第二天的分论坛上我们就有看到生物制品注册分类修订要求的部分内容,
预防和治疗生物制品或将调整为 5 个注册分类类目
,其中治疗生物制品中大家熟知的生物类似物拟将
单独成类
。
仿制药一致性评价——
中国食品药品检定研究院萧红街
萧工从开展一致性评价工作有利于控费和淘汰落后产能和提高仿制药质量展开报告演讲。

中国食品药品检定研究院萧红街工程师
萧工在演讲中提到了一组一致性评价工作的重要数据,截止到 2017 年 5 月 18 号,
完成参比制剂备案的数据记录共 5321 条
,其中属于
289 品种
的备案记录
2882 条
,属于非
289 品种
备案记录
2439 条
,总体来说
289 品种
参比备案记录
更多
,有时间大限的品种中已经有 237 个品种进行了备案登记,52 个暂无备案,其中不乏涉及难以确定参比制剂的品种。
另外,BE 试验由审评制改为备案制以来,目前有新申报的化学药申请 BE 备案 78 件,面向已上市的化学药的一致性评价的 BE 备案 85 件,暂无需要开展临床有效性试验的备案信息。
现场我们也目睹了如果品种通过了一致性评价,即可获得国家版权局批准的“通过一致性评价”标识,将来这两个出现在药品包装盒上的标识将会把那些与原研药临床治疗效果一致的品种和普通仿制药明显
区分
开来。

药品审评改革新趋势——
药审中心黄清竹
黄工的主题演讲,主要介绍了药审中心从 15 年至今,在审评改革工作中做的重要努力。演讲中提到,CFDA
自 2015 年 9 月开始,重点处理药物审评积压问题以来,目前
审评存量已经从 2015 年的 20000 件下降到可控制的 7500 件左右
,工作成效确实显著。

药审中心黄清竹工程师
黄工介绍改革工作进展制度建设时,提到通过建立适应症团队,搭建以临床审评人员为核心,包含药理、毒理、药学、统计等专业审评人员和项目管理人员的适应症审评团队,以更加完善的分工应对未来复杂多样的审评工作。
按照适应症建立审评团队是非常值得期待的审评制度改革,原因在于:
1、闻道先后,术业专攻,专业的人更专注专业适应症,效率更容易提升;
2、制药企业越来越专注特定领域特定适应症的药物研发,企业与适应症审评团队对接沟通,也有利于建立完善的沟通交流机制,符合审评审批改革目的,加快审评速度和提升审评效率。
当黄工报告中提到 60 天临床申请审评时限时,台下听众不约而同的鼓起了掌,足以说明此项政策多么符合制药企业的迫切需求,政策开始执行阶段可能会遇到重重阻力,但是眼下这是一个对于新药研发企业来说绝对利好的政策,其实一个 60 天的审评时限限制,催生出的可能是更加有效的沟通方式,与更快的审评审批效率。通过制度达到审评品种不积压,从提高收费标准开始到现在,CFDA 在不断的前进中促进着中国药物研发的进步。
GCP 和 GMP 检查的现状和挑战——总局食品药品审核查验中心董江萍副主任

总局食品药品审核查验中心董江萍副主任
5 月 24 日,CFDA 发布了《总局关于药物临床试验数据核查有关问题处理意见的公告(2017年第63号)》,关于722 风暴中被检查出有临床试验数据造假行为的企业和医疗机构、CRO 公司的处罚决议尘埃落定,而董主任则在此次演讲中分享了临床试验数据自查核查工作的关键
性数据。
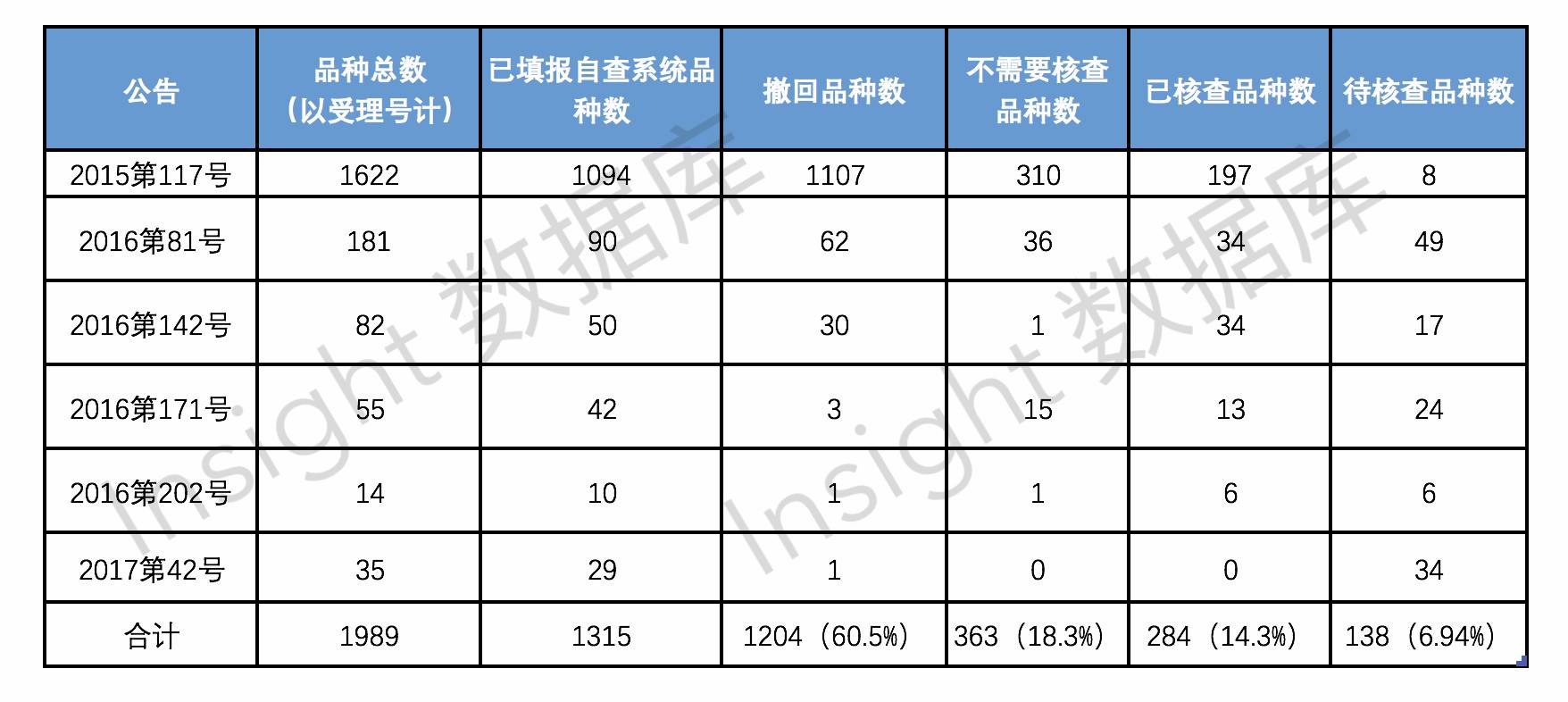 临床试验数据自查核查数据统计
临床试验数据自查核查数据统计
(点击图片可看大图)
如上图所示,截止到 2017 年 5 月,国家食品药品监督管理总局发布了 6 次药物临床试验数据自查核查工作的公告,累计涉及需自查品种 1989 个,撤回品种数 1204 个,目前剩余待核查品种数 138 个,
(注:单位计算均以受理号计)
更多精彩的 CFDA 专场会议 PPT,请在公众号中回复
「DIA」
,即可获得 CFDA 专家的演讲 PPT,今年的政策风向标,就在这里了!

↓ ↓ ↓
点击「
阅读原文
」,免费申请一个 Insight 账号,查询 CFDA 最政策和审评动态数据。



